

The T cell druggable genome
Posted: 3 September 2012 |
The ‘druggable genome’ has been defined as those genes that can be pharmaceutically modulated; when intersected with disease-associated genes, the resultant set represents therapeutic targets for developing drugs to prevent and treat diseases. Historically, druggable therapeutic target genes have been defined by two features; (i) their significant contribution to the disease phenotype in a sufficient number of affected individuals and (ii) modulation of their activity by binding of a drug molecule.
The emerging era of cancer immunotherapy is dramatically changing the target landscape towards molecules that can be recognised by antibodies and T cells, thereby inducing redirection of immune effector mechanisms against target-bearing tumour cells. Despite the fact that the target space for immunotherapy is broad, few genes have been clinically evaluated as cancer immunotherapy targets. We have recently shown that patient and tumourspecific somatic mutations identified by next-generation sequencing of cancer genomes can be systematically targeted by cancer vaccines. In combination with a versatile immunotherapy platform for on-demand production of tailored vaccines, this, for the first time, opens the door to an entirely new source of druggable therapeutic targets – the abundant space of individual cancer mutations.
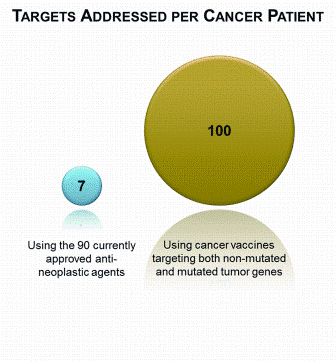
Figure 2: The number of targets addressed for a specific cancer patient. There are 90 currently approved anti-neoplastic drugs, including 9 biologicals2; a patient often receives a chemotherapy regimen comprising three to six chemotherapy agents and a targeted monoclonal antibody (left). A tumour has ~50 immunogenic antigens from protein-changing cancer mutations18,19 and 20-150 gene isoforms with tumour-enhanced expression that, assuming at least one immunogenic epitope per gene, suggests ~50 addressable expression and differentiation antigens (right)
The ‘druggable genome’ has been defined as those genes that can be pharmaceutically modulated; when intersected with disease-associated genes, the resultant set represents therapeutic targets for developing drugs to prevent and treat diseases. Historically, druggable therapeutic target genes have been defined by two features; (i) their significant contribution to the disease phenotype in a sufficient number of affected individuals and (ii) modulation of their activity by binding of a drug molecule.
The emerging era of cancer immunotherapy is dramatically changing the target landscape towards molecules that can be recognised by antibodies and T cells, thereby inducing redirection of immune effector mechanisms against target-bearing tumour cells. Despite the fact that the target space for immunotherapy is broad, few genes have been clinically evaluated as cancer immunotherapy targets. We have recently shown that patient and tumourspecific somatic mutations identified by next-generation sequencing of cancer genomes can be systematically targeted by cancer vaccines. In combination with a versatile immunotherapy platform for on-demand production of tailored vaccines, this, for the first time, opens the door to an entirely new source of druggable therapeutic targets – the abundant space of individual cancer mutations.
The druggable genome: history
The development of small molecule therapeutics typically follows several steps. First, a molecular target, such as a protein, associated with the disease is identified. Second, these proteins are screened to identify those which are druggable, that is those whose function may be modulated by a compound, such as those proteins with a domain, like a binding pocket, accessible to small molecules. The set of druggable genes, the ‘druggable genome’, comprises roughly 3,000 genes, the majority of which code for kinases, receptors, ion channels, proteases, and peptidases1. The number of druggable therapeutic targets, i.e., the intersection between druggable genes and disease genes, is estimated to be 600-1,500. There are 90 currently approved anti-neoplastic drugs2. A cancer patient often receives a chemotherapy regimen comprising three to six chemotherapy agents in addition to surgery and radiation.
In addition to small molecule chemotherapies, a cancer patient will often receive a therapeutic monoclonal antibody (mAbs). The first mAb was first approved for therapeutic use in 1994 and the 24 currently approved mAbs are one of the most successful classes of therapeutic agents2. Antibodies bind proteins on the extracellular surface of cells, where they bind cell markers (e.g., CD20, Her2), receptors (e.g., EGFR) or cytokines (e.g., TNF-α) to block or activate pathways or to label cells for antibody- and complement-dependent cytotoxic elimination by immune cells. Theoretically, the druggable space for mAbs can be any of the 4,080 membrane proteins3. For cancer therapeutics, the druggable therapeutic target space for mAbs is drastically reduced. To exclude toxicities in healthy tissue, the target should be highly upregulated in, or ideally specific to, the cancer cells. In melanoma models, we typically find fewer than 10 membrane proteins that are specific to the melanoma cells, suggesting that the number of the druggable therapeutic mAb targets is of the order 10. An individual cancer patient typically receives one targeted mAb, such as Trastuzumab (Herceptin) targeting the Her2 receptor.
The ‘T Cell druggable genome’
A longstanding vision has been to recruit the immune system’s effectors for cancer therapy by directing them against tumour-specific target molecules. Indeed, the idea of redirecting the immune system to target cancer cells dates to 1890, when Paul Ehrlich proposed generating cancer vaccines. While mAbs, representing the B cell-associated effector molecules of the immune system, are already established as a pharmaceutical drug class, there is growing interest in harnessing the power of cytotoxic T-lymphocytes as therapeutic agents (Figure 1). Indeed, redirecting T cells to combat tumours is a promising therapeutic strategy: T cells have been shown to potently attack cancer cells and induce the rejection of established tumours. Moreover, T cell vaccines are particularly suited for precise targeting of many tumour-associated molecular alterations, including those of intracellular proteins. The first therapeutic cancer vaccine was approved for routine clinical use in prostate cancer in 2011 and further cancer vaccines are in late-stage clinical development4-8. Moreover, clinical testing of adoptive T cell therapies utilising T-lymphocytes with chimeric antigen receptors has shown that the approach can efficiently destroy tumour cells9.
T cell vaccines can be developed against proteins containing a MHC presented peptide. Given that the majority of proteins contain a predicted MHC-presented peptide, the theoretical druggable space for T cell vaccines is huge. On the other hand, a therapeutic target antigen for a cancer vaccine has to fulfil at least two fundamental requirements: (1) immuno – genicity, i.e., a significant antigen-specific immune response must be induced in tumourbearing patients for therapeutic vaccination or healthy individuals for prophylactic vaccination; and (2) selectivity, i.e., the respective epitope is presented mainly or exclusively on patient tumour cells. Over the past two decades, we and others have identified multiple tumour antigens that fulfil these criteria and have experimentally validated them for T cell recognition10,11. Based on defined medical, biological and economic criteria, such as tumour specific expression and penetrance in patient populations, the number of potentially useful cancer vaccine druggable therapeutic targets is fewer than 10012,13. Similarly, using a preclinical melanoma model, we find 32 potential T cell druggable antigens which are significantly over-expressed in the tumour cells compared to healthy tissues.
Alternatively, one could vaccinate against amino acid changes resulting from somatic tumour mutations. Like viral proteins, these mutation antigens are not found in normal cells and thus there would be limited immunological tolerance to them. Accordingly, the mutated epitopes may engage T cells containing highavidity T cell receptors with superior antitumour activity14,15. Indeed, many protein-modifying cancer mutations have been shown to be immunogenic and targeted by T cells16,17. The druggable target space for mutated tumour antigens would then depend on the number of protein-modifying mutations in a tumour. In the melanoma model, there are 962 nonsynonymous point mutations, of which 563 occur in tumour-expressed genes. We tested 50 and found 32 per cent immunogenic, which, when extrapolated to the 563 mutations, suggests 180 immunogenic mutations.
Thus, while the number of non-mutated small molecule and mAb druggable therapeutic targets is limited and a cancer patient will receive three to six chemotherapy agents and a targeted mAb, there are many potential T cell vaccine therapeutic targets. A tumour has ~50 immunogenic antigens from proteinchanging cancer mutations18,19. For a given tumour, 20-150 gene isoforms have tumourenhanced expression (data not shown) that, assuming at least one immunogenic epitope per gene, suggest ~50 addressable antigens. Therefore, the targets in a patient tumour are currently addressed by seven small molecule and mAb therapies while the druggable therapeutic target space for T cell vaccination from both mutated and non-mutated antigens is of the order 100 (Figure 2).
Figure 2: The number of targets addressed for a specific cancer patient. There are 90 currently approved anti-neoplastic drugs, including 9 biologicals2; a patient often receives a chemotherapy regimen comprising three to six chemotherapy agents and a targeted monoclonal antibody (left). A tumour has ~50 immunogenic antigens from protein-changing cancer mutations18,19 and 20-150 gene isoforms with tumour-enhanced expression that, assuming at least one immunogenic epitope per gene, suggests ~50 addressable expression and differentiation antigens (right)
However, while abundant, the tumour mutations are highly patient and tumourspecific, with fewer than five per cent re-occurring in multiple patients20. Therefore, while the ‘T cell druggable genome’ for cancer therapeutics is large, it is highly individualised.
Individualised cancer therapeutics harvest the ‘T cell druggable genome’
The T cell druggable genome for cancer therapeutics is highly individualised. One way to address this large target space is with a therapeutic platform that can generate similarly individualised therapeutics, such as individualised vaccines. To implement such a process for an individualised therapy would require the following steps: identify proteinmodifying somatic mutations in a tumour, prioritise mutations according to their immunogenicity and produce a vaccine targeting these mutations under GMP conditions. Additionally, the whole process should be executed within a short timeframe to account for the aggressiveness of cancer. The IVAC (Individualised VAccine for Cancer) consortium in Mainz, Germany, with the publicprivate partners BioNTech AG, Ribological GmbH and the Institute for Translational Oncology and Immunology (TRON), is leveraging technological advances to make such individualised vaccines.
To efficiently identify the tumour-specific ‘T cell druggable therapeutic genome’, nextgeneration sequencing (NGS) is a game changer. In 2000, the Human Genome Project cost a billion dollars and took 10 years to complete21. Now, the combination of low per-nucleotide costs and higher throughput has resulted in a ‘sequence explosion’ that enables the sequencing of a human genome with coverage of more than 100 fold in a week for a cost of less than USD 10,00022. The current second generation of DNA sequencing platforms begins with short DNA sequences derived from random shearing of the patient tumour and germline genomes. These fragments undergo a local clonal amplification and are processed in a massively parallel platform including several billion parallel sequencing reactions. Next, an extensive bioinformatics pipeline for thorough and fast data analysis and interpretation of the resultant terabytes of data identifies somatic mutations. Thus, the first revolution for individualised vaccines is NGS, including the sequencing platform, extensive laboratory expertise, investments in high-performance computing, robust, highly available mass storage solutions and extensive bioinformatics expertise.
The ability to rapidly generate an individualised vaccine with GMP quality is the second revolution of the IVAC process. Ribological GmbH has developed a production pipeline based on in vitro transcribed RNA (IVT RNA) for cancer vaccines. The RNA-vaccine platform allows rapid production of optimised, highly immunogenic IVT RNA23-26. Ribological GmbH is conducting first-in-man clinical tests using RNA-encoded, non-mutated tumourassociated antigens (TAAs). Furthermore, as this pipeline uses synthesised RNA as a vaccine drug product, it enables the manufacture of cancer vaccine formulations with custom sequences27,28. The whole process takes only weeks, from mutation identification to RNA vaccine availability, and is thus highly suitable for treatment of cancer patients.
In an in vivo study, we demonstrated the proof of concept for the described IVAC process19. By NGS sequencing a murine tumour cell line, we identified 563 mutations resulting in an amino acid change in expressed proteins. Fifty of the mutated sequences were tested for immunogenicity, with one third showing induction of a T cell response in the immunised animals. In tumour transplant models using candidate mutations for peptide vaccination, anti-tumour activity could be shown in prophylactic and therapeutic settings, qualifying the mutations as capable and effective cancer vaccines.
Furthermore, both inter- and intra-tumour mutational heterogeneity can exist in a patient29. The IVAC approach is uniquely positioned to address this heterogeneity as multiple mutations can be simultaneously targeted in a single RNA using an RNA molecule encoding multiple antigens. Mutations can be monitored and detected in the periphery and the IVAC can be updated to improve targeting.
To move IVAC from pre-clinical to clinical testing, the consortium is funded by public and private sources, including the German Federal Ministry of Education and Research ‘Innovative therapies on molecular and cellular basis’ program and the CI3 Excellence Cluster (www.ci-3.de). Regulatory aspects are being collaboratively addressed with the FDA, EMA and Association for Cancer Immunotherapy (CIMT) workgroups to promote ‘actively personalised vaccination’ (APVAC)30.
In summary, the T cell druggable thera – peutic genome is a new target space for cancer drugs. This target space is complementary to both the small molecule and mAb target spaces and, in some cases, much larger. The development of novel ‘on-demand’ therapeutic platforms enables us to effectively address this space. The IVAC approach, with individualised therapeutic RNA cancer vaccines, is a promising approach that will be clinically tested in 2013.
References
1. Hopkins,A.L. and Groom,C.R. (2002) The druggable genome. Nat. Rev. Drug Discov. 1, 727-730
2. Knox,C. et al. (2011) DrugBank 3.0: a comprehensive resource for ‘omics’ research on drugs. Nucleic Acids Res. 39, D1035-D1041
3. (2012) The UnitProt Consortium. Reorganizing the protein space at the Universal Protein Resource (UniProt). Nucleic Acids Res. 40, D71-D75
4. Klebanoff,C.A. et al. (2011) Therapeutic cancer vaccines: are we there yet? Immunol. Rev. 239, 27-44
5. Kantoff,P.W. et al. (2010) Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N. Engl. J. Med. 363, 411-422
6. Kantoff,P.W. et al. (2010) Overall survival analysis of a phase II randomized controlled trial of a Poxviralbased PSA-targeted immunotherapy in metastatic castration-resistant prostate cancer. J. Clin. Oncol. 28, 1099-1105
7. Schwartzentruber,D.J. et al. (2011) gp100 peptide vaccine and interleukin-2 in patients with advanced melanoma. N. Engl. J. Med. 364, 2119-2127
8. Schuster,S.J. et al. (2011) Vaccination with patientspecific tumor-derived antigen in first remission improves disease-free survival in follicular lymphoma. J. Clin. Oncol. 29, 2787-2794
9. Porter,D.L. et al. (2011) Chimeric antigen receptormodified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 365, 725-733
10. Sahin,U. et al. (1995) Human neoplasms elicit multiple specific immune responses in the autologous host. Proc. Natl. Acad. Sci. U. S. A 92, 11810-11813
11. van der Bruggen,P. et al. (1991) A gene encoding an antigen recognized by cytolytic T lymphocytes on a human melanoma. Science 254, 1643-1647
12. Tureci,O. et al. (1997) Serological analysis of human tumour antigens: molecular definition and implications. Mol. Med. Today 3, 342-349
13. Sahin,U. et al. (1997) Serological identification of human tumour antigens. Curr. Opin. Immunol. 9, 709-716
14. Monach,P.A. et al. (1995) A unique tumour antigen produced by a single amino acid substitution. Immunity. 2, 45-59
15. Wolfel,T. et al. (1995) A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science 269, 1281-1284
16. Ikeda,H. et al. (1997) Mutated mitogen-activated protein kinase: a tumour rejection antigen of mouse sarcoma. Proc. Natl. Acad. Sci. U. S. A 94, 6375-6379
17. Lennerz,V. et al. (2005) The response of autologous T cells to a human melanoma is dominated by mutated neoantigens. Proc. Natl. Acad. Sci. U. S. A 102, 16013-16018
18. Segal,N.H. et al. (2008) Epitope landscape in breast and colorectal cancer. Cancer Res. 68, 889-892
19. Castle,J.C. et al. (2012) Exploiting the mutanome for tumour vaccination. Cancer Res. 72, 1081-1091
20. Stratton,M.R. (2011) Exploring the genomes of cancer cells: progress and promise. Science 331, 1553-1558
21. Collins,F.S. et al. (2003) The Human Genome Project: lessons from large-scale biology. Science 300, 286-290
22. (2010) Human genome at ten: The sequence explosion. Nature 464, 670-671
23. Holtkamp,S. et al. (2006) Modification of antigenencoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood 108, 4009-4017
24. Kreiter,S. et al. (2008) Increased antigen presentation efficiency by coupling antigens to MHC class I trafficking signals. J. Immunol. 180, 309-318
25. Kuhn,A.N. et al. (2010) Phosphorothioate cap analogs increase stability and translational efficiency of RNA vaccines in immature dendritic cells and induce superior immune responses in vivo. Gene Ther. 17, 961-971
26. Kreiter,S. et al. (2010) Intranodal vaccination with naked antigen-encoding RNA elicits potent prophylactic and therapeutic antitumoral immunity. Cancer Res. 70, 9031-9040
27. Pascolo,S. (2006) Vaccination with messenger RNA. Methods Mol. Med. 127, 23-40
28. Kreiter,S. et al. (2011) Tumour vaccination using messenger RNA: prospects of a future therapy. Curr. Opin. Immunol. 23, 399-406
29. Gerlinger,M. et al. (2012) Intratumour heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 366, 883-892
30. Fox,B.A. et al. (2011) Defining the critical hurdles in cancer immunotherapy. J. Transl. Med. 9, 214
About the authors
Dr. Sebastian Kreiter is an expert in vaccine development and T cell immun – ology. He is a trained physician and specialist in internal medicine. In 2001, he joined the team of Ugur Sahin and Özlem Türeci and made key contributions to the development of the optimized RNA based cancer vaccine approach. He is the co-inventor on several patent applications. Currently, he is heading the Immunotherapy Development Center within TRON, where he is responsible for all non-clinical efficacy testing, mode-of-action studies and early development projects.
Dr. Martin Löwer received his PhD in Bioinformatics at the Goethe University Frankfurt, applying structure based virtual screening to target human pathogens. His areas of expertise include mathematical classification methods and bioinformatics software development and optimisation. He has developed support-vector machine (SVM), self-organising maps (SOM), neural nets, nextgeneration sequencing and random forest algorithms for cancer and infectious disease drug development.
Dr. John Castle originally trained in physics at the University of Washington and at MIT, developing advanced inverse theory methodologies to image deep earth structure. For the next 10 years, he worked at Rosetta Inpharmatics, part of Merck Sharp and Dohme (MSD), developing novel biotechnologies for drug development. In Mainz, he is Co-Director of the Biomarker Development Center and combines immunology, computational biology, and genomics for the development of individualised cancer immunotherapies.
Dr. Jan Diekmann studied Biochemistry at the Eberhard-Karls-University Tübingen and completed his PhD in Immunology at the University Clinic Tübingen and University Clinic Würzburg. To improve immunotherapy against Epstein-Barr virus-associated malig – nancies, he investigated the antigen-processing pathways of Epstein – Barr virus proteins and the generation of EBV-specific T cell lines. In 2009, he joined Ugur Sahin and Özlem Türeci to work on the development of the T cell vaccine platform.
Dr. Özlem Türeci is an MD by training. Her research focus is the identification of immunotherapeutic drug targets and development of immunotherapeutics against cancer. She is co-founder and Chief Executive & Medical Officer of Ganymed Pharmaceuticals, a venture capital-backed biotech company based in Mainz/Germany. Moreover, she is chair and co-initiator of the Cluster of Individualised Immunointervention (CI3), a winner of the German leading edge Cluster competition.
Professor Dr. Ugur Sahin is the CEO of BioNTech, leads an independent oncology research team at the Johannes- Gutenberg University Mainz and is the founder and director of TRON, Translational Oncology at the Mainz University Medical Center. He is develop – ing novel cancer vaccines and individualised cancer immunotherapies. His current research includes the identifica – tion and characterization of new tumour antigens, the development of RNA-based cancer vaccines and the development of diagnostic tools for the early detection of cancer.
