

Future proofing risk assessment: preventing the next nitrosamine incident
Posted: 17 December 2021 | Dave Elder (David P Elder Consultancy) | No comments yet
The valsartan nitrosamine contamination issue was probably the most significant quality issue to hit the pharmaceutical industry in a decade. Here, Dave Elder outlines how agencies and manufacturers are dealing with the problem and indicates the knowledge gaps that might be concealing future complications.
THE NITROSAMINE contamination issue was first identified in July 2018 and has resulted in thousands of recalls, millions of dollars of lost sales and untold reputational damage. There was huge regulatory concern because N-nitrosamines are potential human carcinogens and are toxic at extremely low levels, ie, ng/day levels. Initially, the issue was restricted to N-nitrosodimethylamine (NDMA) contamination of valsartan from Chinese active pharmaceutical ingredient (API) suppliers. However, it soon became apparent that use of other common secondary or tertiary amines in API syntheses could also introduce other types of nitrosamine impurities besides NDMA. These N-nitrosamines include NDEA (N-nitrosodiethylamine), NMBA (N-Nitrosodibutylamine), NMPA (N-Nitrosomethylphenylamine), NIPEA (N-Nitrosoisopropylethylamine) and NDIPA (N-Nitrosodiisopropylamine). Additionally, other sartan drug products containing a tetrazole ring – eg, candesartan, irbesartan, etc – from different manufacturers in China, India and further afield were also found to contain unacceptable levels of nitrosamines.1,2
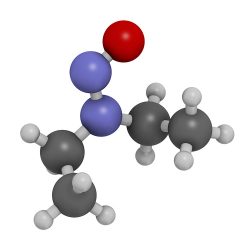
N-Nitroso-diethylamine (NDEA)
In September 2019, some common dyspepsia products (ranitidine and nizatidine) were found to contain unacceptable levels of NDMA. It was subsequently identified that NDMA levels in some ranitidine products stored at ambient temperature can increase over time. By Spring 2020, global agencies were requesting permanent withdrawals of these products from their markets.1,2
In December 2019, global agencies became aware that some metformin products had elevated levels of NDMA. Metformin extended‑release products were found to be implicated, while the input API and metformin instant-release products were not affected, suggesting that degradation rather than contamination were the causative factors.1,2
In the latter half of 2020, a new class of drugs – rifamycin antibiotics, ie, rifampicin and rifapentine – were implicated and a further two, novel N-nitrosamines – MNP (1-methyl-4- nitrosopiperazine) and CPNP (1-Cyclopentyl-4- Nitrosopiperazine) – were found.1
Finally, several drugs were identified as containing API-nitrosamines, ie, N-nitrosovarenicline (varenicline)3 and N-nitrosoirbesartan (irbesartan),4 leading to product recalls.
Root causes for the presence of nitrosamine impurities in APIs
By mid-2020, the likely root causes of nitrosamine formation were well established, at least for short‑chain alkyl nitrosamine contamination in sartans, ie, NDMA and NDEA. Formation of short‑chain alkyl nitrosamines is likely in the presence of secondary, tertiary or quaternary amines and nitrite salts at pHs below seven. Under these acidic conditions, nitrite salts can form nitrous acid, which in turn can react with these short‑chain alkyl amines, forming the corresponding nitrosamine. There is a significant risk of nitrosamine formation if nitrous acid is used to quench residual azide, a reagent commonly utilised in tetrazole ring formation or the introduction of azide functional group into sartan molecules, in the presence of precursor short-chain alkyl amines.1,2
By mid-2020, the likely root causes of nitrosamine formation were well established”
Starting materials, intermediates and indeed the API – eg, irbesartan (and specific API degradants) – may all contain secondary or tertiary amine moieties. Reagents or catalysts can also include tertiary or quaternary amine functionalities. In addition, common amide solvents such as N,N‑dimethylformamide, N-methylpyrrolidone, N,N‑dimethylacetamide and N,N-diethylacetamide can all contain secondary amines as impurities.1,2
Nitrosamine impurities can also be introduced when vendor-sourced materials, including starting materials and raw materials, are contaminated with nitrosamines. Recovered solvents, reagents and catalysts may also pose a significant risk of nitrosamine impurities. Some drug products using APIs, eg, pioglitazone, manufactured by certain ‘low risk’ processes, were found to be contaminated with nitrosamines.1,2
Another possible source of nitrosamine contamination is lack of appropriate optimisation of the API manufacturing process in those cases when reaction conditions and addition-order of reagents, intermediates or solvents are poorly controlled. As such, multiple strategies may be necessary to identify all potential sources of contamination.1,2

Finally, different mechanisms may be in play for drug products compared to APIs. For instance, nitrites are common nitrosating impurities that have been reported in many excipients at ppm levels;5 these may lead to nitrosamine impurities forming in drug products during the drug product manufacturing process and shelf-life-storage period. Manufacturers should also be mindful that nitrite and nitrosamine impurities may be present in potable water. Some drug products may undergo degradation pathways that form nitrosamine impurities, eg, ranitidine, varenicline and irbesartan; which could potentially occur during drug product storage.1-4
Risk assessments
Regulatory agencies were quick to realise the potential implications on global supply chains and requested that manufacturers consider the potential causes of nitrosamine formation in all APIs and drug products; not just sartans, ranitidine and metformin extended-release products, etc.1,5
If a risk of nitrosamine impurities is identified, confirmatory testing of batches should be conducted using sensitive and appropriately validated methods, ie, HPLC-MS-MS or GC-MS-MS.1 If the risk assessment determines that there is no potential for nitrosamine impurities, there is no need for further action. If a nitrosamine impurity is detected, API manufacturers should investigate the root cause (see previous section). They should implement changes in the manufacturing process to reduce or prevent nitrosamine impurities.
Additionally, for those processes which are sub-optimal, different failure modes could result in different nitrosamines in different amounts across different batches utilising the “same process” and the same API producer, with contamination detected in some batches but not all. This must be taken into consideration in the risk assessment.1
Future proofing risk assessment

Is there anything that industry/regulators can do to prevent the next nitrosamine‑like contamination or impurity issue? This is a really difficult question to answer, but the most likely answer is no. This is because risk assessment is based on the likelihood of a risk arising, ie, prior knowledge, and therefore, if the risk has never been encountered before then this potential risk will always be scored low on any risk-matrix. Once the underlying knowledge is available then that risk can be scored appropriately. Many commentators were highly critical that the original risk assessment conducted by Chinese API suppliers did not identify the risk of potential nitrosamine formation.5 However, risk is frequently in the eye of the beholder, especially when a significant process-chemistry risk masks another underlying potential risk. The valsartan contamination problem “occurred because NaNO2 was used as an azide quenching agent to address the significantly more hazardous, in the view of the risk assessors, formation of hydrazoic acid, which poses a dangerous explosive risk when shocked or heated”.5 This view, that the risk was not immediately apparent, was also shared by the US Food and Drug Administration (FDA). They indicated in early 2019 that, although they had issued a recent guidance on mutagenic impurities risk assessments in March 2018, “neither regulators nor industry fully understood how NDMA and NDEA could form during this particular manufacturing process”.6
When significant understanding of how nitrosamines form was achieved, it became appropriate for global regulators to ask industry to perform robust risk assessments that could be used to either discharge the risk or perform analysis to confirm/discharge that risk. However, FDA has indicated that “Despite the nearly two and a half years into this contamination issue, we have still many ongoing challenges”.7 Additionally, manufacturers and regulatory agencies are still “being surprised quite frequently” by the results arising from testing their products, another FDA spokesperson indicated.5 These cases include European regulators recalling batches of sartans due to the presence of azido impurities in August 2021.8 This is surprising, given that this azide chemistry is commonly utilised in tetrazole ring formation or the introduction of azide functional group into sartan molecules.

Additionally, Pfizer expanded its voluntary recall of Varenicline (Chantix) to include all lots of the product.3 It recalled these lots due to the presence of unacceptable levels of N-nitroso-varenicline. Lupin also recalled batches of irbesartan tablets and irbesartan and hydrochlorothiazide tablets due to the presence of nitrosoirbesartan.4 Thus, although there was significant understanding of the formation of short alkyl chain nitrosamines, such as NDMA, NDEA, etc, formation of higher molecular weight nitrosamines can involve different mechanisms including nitrite in excipients and water. An FDA spokesperson recently indicated that “other routes of formation have also been found, including some that, like malting, involve extracting moisture”. She further indicated that “we still truly have an incomplete understanding of all the possible ways these impurities could form”.7
The most significant challenge is a lack of understanding and knowledge surrounding by-product chemistry, ie, impurities and degradants. Despite our knowledge and understanding of process chemistry and related impurities being well established from an ICH Q3A(R2) and ICH Q3B(R2) perspective – ie, 0.1 percent (= 1,000 ppm), in contrast, the myriad unusual chemical reactions that can occur leading to very small quantities of unknown impurities at ppm and particularly ppb levels, are generally not well known or researched. As such, risk assessments based on an incomplete knowledge rarely highlight all possible risks.
About the author
Dave Elder has nearly 40 years of service within the pharmaceutical industry at Sterling, Syntext and GlaxoSmithKline. He is now an independent GMC consultant. He is a visiting professor at King’s College, London and a member of the British Pharmacopoeia. He is a member of the Joint Pharmaceutical Analysis Group (JPAG) and the Analytical Division Council of the Royal Society of Chemistry.
References
- FDA. 2021. Control of Nitrosamine Impurities in Human Drugs − Guidance for Industry R1, FDA CDER CGMP. https://www.fda.gov/media/141720/download. Accessed 08 November 2021.
- EMA. Lessons Learnt from Presence of N-nitrosamine Impurities in Sartan Medicines. 2020. https://www.ema.europa.eu/en/documents/report/lessons-learnt-presence-n-nitrosamine-impurities-sartan-medicines_en.pdf. Accessed on 08 November 2020.
- Edney A. Carcinogens Still Vex Drug Industry Years After Recalls Began. 01 July 2021. Bloomberg. https://www.bloomberg.com/news/articles/2021-07-01/carcinogens-still-vex-drug-industry-years-after-recalls-began. Accessed on 08 November 2021.
- Nationwide Recall of All Irbesartan Tablets and Irbesartan and Hydrochlorothiazide Tablets Due to Potential Presence of Impurity. 19 October 2021.
- Elder DP, Johnson GE, Snodin DJ. Tolerability of risk: A commentary on the nitrosamine contamination issue. J. Pharm. Sci. 2021 Jun;110(6):2311-2328.
- Gottlieb S, Woodcock J. FDA Statement on the FDA’s Ongoing Investigation Into Valsartan and ARB Class Impurities and the Agency’s Steps to Address the Root Causes of the Safety Issues. 25 January 2019a. https://www.fda.gov/news-events/press-announcements/fda-statement-fdas-ongoing-investigation-valsartan-andarb-class-impurities-and-agencys-steps. Accessed on 08 November 2021.
- Nitrosamines as Impurities in Drugs; Health Risk Assessment and Mitigation Public Workshop. 29-30 March 2021. https://www.fda.gov/drugs/news-events-human-drugs/nitrosamines-impurities drugs-health-risk-assessment-and-mitigation-public-workshop-03292021. Accessed on 08 November 2021.
- Risk of the presence of mutagenic azido impurities in losartan active substance. 29 September 2021. https://www.edqm.eu/en/news/risk-presence-mutagenic-azido-impurities-losartan-active-substance. Accessed on 08 November 2021.
Issue
Related topics
Active Pharmaceutical Ingredient (API), Drug Manufacturing, Drug Safety, Formulation, Good Manufacturing Practice (GMP), ICH guidelines, Impurities, Industry Insight, Regulation & Legislation, Therapeutics
Related organisations
Related drugs
Candesartan, Irbesartan, metformin, nizatidine, ranitidine, rifampicin, rifapentine, Varenicline, Varenicline (Chantix)





