Characterising therapeutic antibodies and ADCs using mass spectrometry
Posted: 24 August 2016 | Ioannis A. Papayannopoulos (Celldex Therapeutics Inc.) | No comments yet
Since the 1970s, the advent of biotechnology has resulted in the development and commercialisation of many therapeutic proteins, including antibodies and antibody fragments, for the treatment of human diseases. Examples include antibody treatments for autoimmune diseases (for example, adalimumab [Humira®] for rheumatoid arthritis), cancers (such as trastuzumab [Herceptin®] for breast cancer), degenerative conditions (including ranibizumab [Lucentis®] for macular degeneration), viral and bacterial infections, as well as others. Additionally, antibodies are used as imaging reagents, linked to radionuclides or non-radioactive rare earth metals, and quite extensively employed as reagents in biomedical research, commercial medical tests and clinical laboratory assays (for example, enzyme-linked immunosorbent assay [ELISA]).
Therapeutic antibodies are typically monoclonal antibodies (MABs) of the immunoglobulin G (IgG) isotype, with affinities against specific antigens and which bind monospecifically to particular cells or proteins. They work either by stimulating the patient’s immune system, thus priming or enhancing the immune response, or by binding to specific molecules and thus modulating or inhibiting certain biochemical pathways involved in disease. Monoclonal antibodies have also been developed with affinities towards specific tumour antigens to deliver radionuclides to them, thereby killing tumour cells with minimal deleterious effects on non-tumour cells. Such radiommunotherapeutic antibodies are modified with a chelator that binds a radioactive metal ion that is thus transported to the tumour.
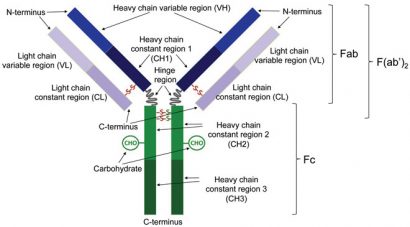
Figure 1: Schematic representation of an antibody. Enzymatic cleavage at the hinge region results in the generation of heavy and light chains that are linked via a disulfide bond (Fab). Enzymatic cleavage below the disulfide bonds that link the heavy chain constant regions 2 results in the generation of a fragment consisting of a pair of disulfide-linked heavy and light chains that are also linked to one another (F(ab’)2)
Antibody-drug conjugates (ADCs), used in cancer chemotherapy, have small-molecule chemotherapeutics covalently attached via a linker that is cleaved at the tumour site, thereby releasing a cytotoxic small molecule in the immediate vicinity of a tumour. Although only a few ADCs have received regulatory approval so far, they comprise a very active area of research and development in many biopharmaceutical companies.
Structure of therapeutic MABs and ADCs
An IgG therapeutic antibody comprises two identical longer sequences (heavy chains) and two, also identical, shorter protein sequences (light chains). IgG light chains are typically 211-217 amino acids long, whilst IgG heavy chains comprise approximately 450 amino acids. The two heavy chains are linked covalently with disulfide bonds between cysteines roughly in the middle of the respective sequences, in a Y-shaped configuration (Figure 1). The number of disulfides linking the two heavy chains depends on the IgG subclass (IgG1, IgG2, IgG3 and IgG4 isotypes). Each light chain is covalently attached to a heavy chain with a single disulfide bond between cysteine residues. Along with interchain disulfide bonds, each heavy and light chain also has several intra-chain disulfide bonds between its cysteine residues.
IgGs are N-glycosylated at a single site on each heavy chain. In addition to the covalent links between cysteines, the antibody chains are also held together by electrostatic and hydrophobic interactions. The molecular mass of each light chain is approximately 23 kDa and that of each heavy chain is around 52 kDa, making the total molecular mass for the intact antibody approximately 150 kDa.
Each chain is divided into domains of approximately 110 amino acids; thus each light chain has two domains and each heavy chain four. The domain at the N-terminus of the heavy chain and the domain at the N-terminus of the light chain are called the variable region and together they comprise the part of the antibody that recognises and binds to a specific antigen. The C-terminal domain on the light chain and the other three domains on the heavy chain are called the constant regions and their amino acid sequences are usually identical in all antibodies of the same isotype of a particular organism.
An ADC is made by modifying a MAB that recognises an antigen associated with a particular tumour. A small-molecule chemotherapeutic, known to be effective against the particular tumour but often too toxic to be administered via standard chemotherapy (such as the antineoplastic agent monomethyl auristatin), is conjugated to the IgG with a linker that is cleavable by enzymes present in the immediate vicinity of the tumour. Thus, the cytotoxic molecule is delivered to and released at the tumour site with minimal damage to healthy cells and organs. The small molecule and the cleavable linker are conjugated to the IgG via cysteines thiol after partial disulfide reduction or to the ε-amines on lysine residues. Typically, there is a distribution of such small molecules added to a MAB and this is reflected in the change in molecular mass (Figure 2).
Characterisation of antibodies
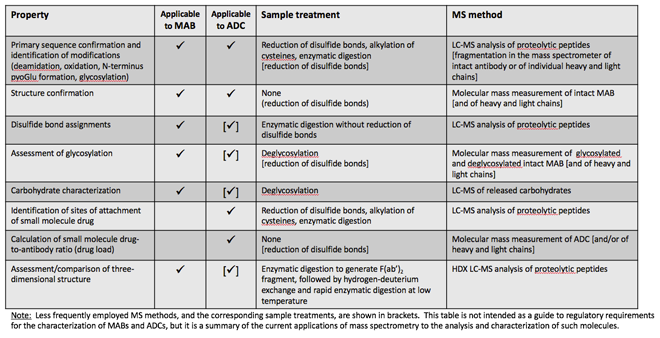
Of all analytical techniques applied to the characterisation of MABs and ADCs, liquid chromatography coupled to mass spectrometry (LC-MS) is arguably the most powerful and capable of providing detailed characterisation down to the level of individual amino acids. Mass spectrometric methods for the characterisation of antibody and protein pharmaceuticals have been developed and used since the 1980s. They have been continuously refined, keeping pace with the significant improvements during this time in the performance (speed, sensitivity, resolution and software) of chromatography and MS instruments. The broad categories of characterisation of MABs using MS are summarised as follows.
Primary sequence confirmation
This is the earliest application of MS for MAB characterisation. An antibody is digested, typically after reduction and alkylation of cysteines, with an enzyme of known and reproducible specificity. Proteolytic peptides thus generated are separated by high-performance liquid chromatography (HPLC) or, increasingly during the past decade, by ultra-HPLC (UPLC) inline with MS; this is referred to as a ‘peptide map’ and the overall approach as ‘bottom up’ LC-MS characterisation. Molecular masses, as well as fragmentation mass spectra of pep tides, are measured and matched to the known sequence of the recombinant MAB. The preferred enzyme for digestion is trypsin, which cleaves at the C-terminus of lysine and arginine residues. These amino acids tend to be fairly evenly distributed in most MABs, indeed in most proteins, and the majority of proteo lytic peptides are 8-30 amino acids long, which is the optimal size for most mass spectro meters. The mass spectrometric data thus generated can account for 70-80% of the MAB sequence.
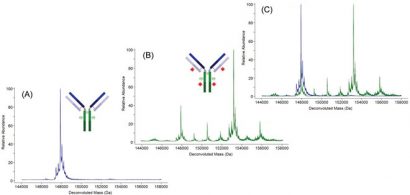
Figure 2: Deconvoluted molecular mass spectra of an antibody (A), the same antibody after conjugation to a small molecule drug (B), and of the two molecular mass spectra superimposed (C)
Usually digestion with a single enzyme such as trypsin does not yield full sequence coverage because some very small and hydrophilic peptides, or some very large peptides, are also produced; the former may not be retained on a reversed phase HPLC column, whilst the latter may not ionise with adequate efficiency in some mass spectro meters. At an early (preclinical) stage of development of a therapeutic MAB, a single enzyme digest that yields coverage of most of the amino acid sequence is quite adequate. Later on, accounting for every single amino acid in the sequence becomes necessary and digestions with additional enzymes of different specificities than trypsin are carried out to obtain information from the regions of the sequence that failed to be identified in the tryptic peptide map.
With some high resolution mass spectro meters, good fragmentation data can be obtained from the intact heavy and light chains, or from fragments thereof, generated with specific enzymes (Figure 1). These data are used to confirm the amino acid sequence and also pinpoint known modifications. This approach, which is gaining traction in MAB characterisation, is referred to as ‘top down’ or ‘middle down’ LC-MS.
With ADCs the same considerations apply, although the small molecule introduces certain complications. Enzyme cleavage sites available in the MAB may be blocked in the ADC when the small molecule is conjugated to the lysine side chains, where trypsin can no longer cleave. Furthermore, some small molecules are especially hydrophobic, thus affecting chromatographic separation. Therefore, a particular enzyme digestion procedure that was pains takingly developed and optimised for the MAB may not work very well for the ADC.
Identification and characterisation of post-translational modifications
Therapeutic antibodies have modifications to their amino acid sequence, some of which, such as glycosylation, which will be discussed in more detail in this article, occur during the process of protein production in cell culture. Other modifications are artifacts introduced during the purification and formulation process, during storage, or at times by the analytical methods employed. Such modifications include: formation of pyroglutamate when a glutamine or glutamic acid is present at the N-terminus of the MAB; oxidation of susceptible amino acid side chains (usually methionine); and deamidation of asparagine side chains with conversion to aspartic acid. This is by no means an exhaustive list, with new and unusual modifications to MABs being reported regularly.
Such modifications can be identified and characterised by MS from the measured molecular masses and the fragment ion mass spectra of the peptides containing the modified amino acids. When a modification is only partial some rough quantitation may be obtained from the relative abundances of the respective peptide ions.
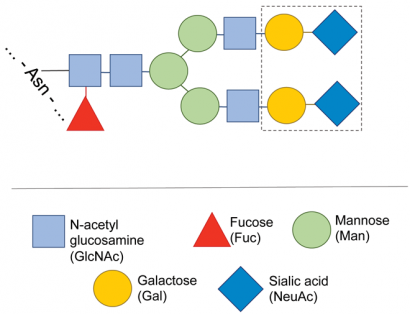
Figure 3: Typical structure of the carbohydrate of a monoclonal antibody. The carbohydrate heterogeneity usually observed in MABs is due to the presence of a mixture of carbohydrate structures, missing one or more of the sugars shown inside the dotted boundary
In ADCs, the conjugation of the small molecule to specific amino acids is a modifica – tion that is introduced deliberately to the MAB. The peptide map data can be used to identify amino acids thus modified. The process of conjugation may also introduce modifications when the conjugation chemistry takes place under conditions that are not optimal to a MAB.
A special type of modification is the formation of disulfide bonds. In MABs disul fides form between cysteines on the same chain and between cysteines on different chains. Characterisation of disulfide bond connectivity in proteins can be complicated, and this is especially true for MABs, which have many cysteines (32 in IgG1, 36 in IgG2). It is achieved by digestion of the MAB without prior reduction of cysteines and assignment of disulfides using MS data. This is far from trivial an exercise, as conditions that maintain the integrity of disulfide bonds (such as a low pH) are often suboptimal for most specific enzymes that are typically used for digestion.
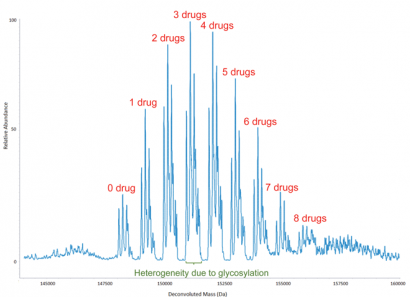
Figure 4: Deconvoluted molecular mass spectrum of an antibody-drug conjugate (ADC), which shows the distribution of small-molecule drugs attached to antibody molecules. The overall drug-to-antibody ratio is calculated as the relative abundance weighted average of the individual ADC peaks. It is worth noting that the heterogeneity due to glycosylation is reflected, as would be expected, not only in the peak with no drug attached but also in each of the ADC peaks
Characterisation of glycosylation
Antibodies are usually glycosylated with a complex N-linked carbohydrate attached to an asparagine residue in the constant region of the heavy chain. Less frequently, O-linked glycosylation may also be present. An overall assessment of glycosylation can be made from the MS measurement of the molecular mass of the intact antibody or, after disulfide reduction, of the heavy chain. The difference between the measured molecular mass and that calculated from the known MAB sequence is the mass of the carbohydrate. Since in most MABs the carbohydrate structure is fairly standard (Figure 3), it is often possible to calculate the carbohydrate composition from this mass difference. The same information can also be obtained from the peptide map, from the glycosylated peptides generated by enzymatic digestion.
More definitive information is obtained by enzymatically releasing the carbohydrate from the antibody and performing characterisation separately. There are several methods employed for the analysis of carbohydrates that do not involve MS. However, MS is increasingly being used to characterise protein carbohydrates. Chemical derivatisation methods have been developed to make carbohydrates amenable to reversed phase HPLC, thus enabling the use of the same instruments for their analysis as those employed for the analysis of peptide maps. Whilst molecular masses may account for the sugar composition, fragmentation in the mass spectrometer allows for determining the connectivity of the various sugars comprising the MAB carbohydrate.
Determination of the drug-to-antibody ratio in ADCs
The amino acids of a MAB to which a small-molecule drug has been conjugated can be identified from the peptide map of the ADC. Typically, there is a distribution of small molecules attached to a MAB, complicating calculations of the drug-to-antibody ratio. To obtain information on the distribution and the average number of small molecules conjugated to an antibody, the molecular mass of the latter needs to be measured (Figure 4), from which the extent of conjugation and its distribution may be determined. The overall drug-to-antibody ratio (drug load) is calculated as the weighted average of the individual ADC peaks. This is an important measurement since it reflects the reproducibility of the manufacturing process for the ADC and, of course, the drug load impacts the therapeutic properties of the ADC.
Hydrogen-deuterium exchange MS
The tertiary structure of antibodies can be probed by a number of physicochemical methods, such as X-ray crystallography, differ ential scanning calorimetry and spectroscopy. A very powerful mass spectrometric approach, hydrogen-deuterium exchange mass spectrometry (HDX-MS), is being used with increased frequency to assess the tertiary structure of proteins, including antibodies. Briefly, an antibody, or the F(ab’)2 fragment thereof, that contains the two linked heavy and light chain pieces (see Figure 1) and which can be generated by the use of specific enzymes, is incubated in deuterated water, leading to the exchange of hydrogens of amide bonds with deuterons from the solvent. This process is very sensitive to solvent accessibility and structure flexibility, and only hydrogens on those amides that are exposed to the solvent are exchanged.
The hydrogen-deuterium exchange is quenched by lowering the pH, followed by rapid digestion with pepsin and rapid UPLC separation at low temperature to minimise the back exchange of deuterium with hydrogen, with on-line mass spectrometric analysis. Timing and standardisation of all steps are critical for reproducibility, which is achieved with cooled automated liquid handling equipment. HDX-MS data are processed and presented in a readily interpretable format with specialised software.
Identification of peptides that exhibit significant deuterium incorporation and those that exhibit little or none can be used to determine which regions of the sequence are exposed to the solvent and which are shielded. This information is correlated to protein folding and used to assess aggregation, compare different batches or, indeed, different antibodies of the same isotype such as biosimilars, as well as to carry out epitope mapping and ligand binding studies. The advantage of HDX-MS over X-ray crystallography is that the former requires significantly less material, is much faster and can tolerate impurities.
Conclusion
MS is indispensable in the analysis and characterisation of recombinant protein pharmaceuticals such as MABs and ADCs, as summarised in Table 1 (page 61). Of course MS, typically in combination with on-line liquid chromatography, is also indispensable in discovery research and in clinical research applications involving MABs and ADCs. In the development of antibody drugs MS is used to generate detailed and definitive information on their primary structure; identify and characterise expected and unexpected modifications; provide qualitative and quantitative measurements of conjugated small molecule drugs in ADCs; characterise carbohydrate glycoforms; and offer insights on the tertiary structure and folding of these large and often complex molecules.
About the author
Ioannis Papayannopoulos has been carrying out peptide and protein analysis and characterisation work, using mass spectrometry, chromatography and other analytical techniques, for over a quarter of a century. He received his undergraduate degree in Chemistry from Bowdoin College and his PhD in Organic Chemistry from the Massachusetts Institute of Technology, where he conducted research in mass spectrometry under the supervision of the late Professor Klaus Biemann. He has held senior scientific and management positions in the biopharmaceutical industry, at such companies as AstraZeneca, Biogen, EMD Pharmaceuticals and Targanox, and in academia, most recently as the Director of the proteomics core facility at the Koch Institute for Integrative Cancer Research at MIT. Currently he is with Celldex Therapeutics, a biotechnology company in Massachusetts, working on the analysis and characterisation of antibodies, antibody-drug conjugates and recombinant protein pharmaceuticals.