Quality control of freeze-dried oral formulations; challenges when developing novel approaches for the delivery of poorly-soluble drugs
Posted: 22 August 2017 | Abdulmalik Alqurshi, Paul Royall | No comments yet
A common obstacle encountered in the early stages of drug development is the formulation of poorly water-soluble drugs (PWSDs). One effective approach to improve the dissolution of PWSDs is to render such drugs into their amorphous or disordered form. However, amorphous materials are both physically and chemically unstable, and tend to revert back to their crystalline state when exposed to the physical and thermal processes that are typically involved in the production of oral dosage forms. The authors have developed a novel formulation approach based on directly freeze-drying a solution of a PWSD held in a gelatin capsule. Such an in-situ approach circumvents processing instability issues and unexpectedly led to a marked increase in the dissolution of nifedipine (t1/2 = 1.88 ± 0.05 minutes) when compared to the equivalent marketed product. In this article we discuss the quality control challenges and how quality control was applied to our novel amorphous formulation platform…

DRUG candidates that show poor solubility and low dissolution rates are a frequent challenge for the pharmaceutical industry, as expensive strategies are required to overcome this limiting oral bioavailability.1-4 A solubility of >60μg/mL is a typical target for developable drugs, as this solubility usually leads to a satisfactory absorption provided the drug has acceptable permeability.5
Improving a drug’s kinetic solubility by means of dissolution rate enhancement techniques is typically based on the governing factors of the Noyes-Whitney equation.1,2,6 One strategy is to increase particle surface area using processing techniques such as spray drying or milling. Such techniques exert physical and thermal stress and therefore are unsuitable for thermally sensitive drugs.7-10 An alternative is to render a PWSD into its amorphous state by creating a molecular dispersion of the drug in a water-soluble carrier.2,6,11 However, formulating amorphous materials is a challenge, as they are thermodynamically metastable12 and will revert back to their crystalline form when stored at a temperature where nucleation is thermodynamically and kinetically favourable.13,14
Crystallisation is influenced by other environmental conditions, such as humidity. Thus, poor environmental control will add risk to the manufacturing, processing, packaging and storage of amorphous medicinal products. The chemical instability of drugs in their amorphous form is also reported to be higher when compared to their crystalline equivalents.15,16 This has been attributed to the enhanced level of molecular mobility observed in amorphous materials. Although amorphous systems are not new to the pharmaceutical field, the strategies employed to increase stability and the analytical tools required to measure the success of these approaches vary considerably.11,17 The selection and development of robust quality control procedures for amorphous pharmaceutical products is therefore difficult to achieve, and a range of methods are usually deployed.
One parameter that is nearly always measured in amorphous materials is the glass transition temperature (Tg).17 When an amorphous material is heated, the hard rigid glass converts into a less viscous supercooled liquid. The midpoint in this process is the Tg, and above this temperature the higher mobility within the system permits a much faster rate of crystallisation and chemical degradation.18,19 Rapidly heating and cooling a mixture of a drug in the presence of a polymer will usually result in a glassy solid solution; hot melt extrusion is based on this approach. If thermal stability of the drug under development cannot be assumed, alternatives are required. One approach is the rapid removal of a solvent from a solution containing the drug – with limited time to permit nucleation and crystal growth, a disordered or amorphous material results.
Freeze drying is based on solvent removal – first by freeze concentration, followed by sublimation, and then finally by evaporation.20 The authors’ article in Issue 3 of European Pharmaceutical Review gives a succinct overview of the freeze-drying process.21 Formulations produced by this method are highly porous, low-density solids, which are extremely fragile, making secondary processing difficult. Thus, the majority of freeze-dried medicinal products are formed in-situ within their primary packaging, eg, blister packs or ampoules.22 The successful Zydis platform technology is a good example of where the dosage form, in this case an orally disintegrating tablet, is freeze-dried in-situ within its blister pack.

Figure 1: Top left clear gelatin capsules filled with TBA based feed solution of nifedipine, bottom left, Red opaque gelatin capsule to prevent light induced degradation of nifedipine. Photo on the right; Scanning electron microscope (SEM) of freeze-dried capsule bottom and non-freeze dried capsule top, no damage is observed in the structure of the freeze dried capsule bottom.
This approach is often used for the formulation of poorly soluble compounds; for example, loratadine. A novel but logical progression of this in-situ approach is the direct freeze-drying of a drug-containing solution preloaded in a standard gelatin capsule. This will allow small batches of a novel amorphous formulation to be prepared and tested, which is useful for when limited amounts of the drug are available early in the development cycle. The authors have recently prepared an in-situ freeze-dried capsule formulation for the insoluble drug nifedipine (Figure 1). Surprisingly, the current marketed capsule’s dissolution rate was three times slower than our capsule – 80% of the 10mg dose of nifedipine was dissolved within six minutes from the in-situ freeze-dried capsule (Figure 2).
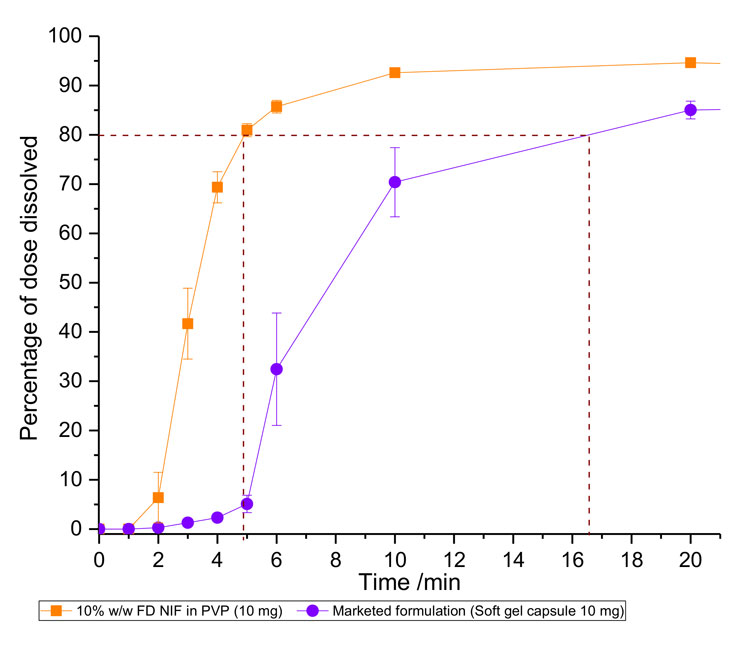
Figure 2: Dissolution profile of the TEVA soft gel 10mg nifedipine capsule and our novel in-situ freeze-dried capsule (purple), containing 10mg of nifedipine dispersed with PVP to form a 10% w/w solid amorphous solution (orange). Average T80 for the marketed formulation is approximately three times longer than that of the in-situ capsule FD formulation (10% w/w NIF in PVP). Error bars represent standard error of n=3.
The specific hydrogen-bonding interactions of nifedipine with the water-soluble polymer polyvinylpyrrolidone (PVP) and the high porosity of the freeze-dried cake within the capsule were the mechanistic basis for this improvement in dissolution.23 The marketed formulation was a liquid-filled capsule and, upon disintegration of the capsule, a dispersion of oil/lipid droplets formed. Thus, the dissolution data indicates that the nifedipine from the marketed formulation is retained within these droplets; however, when presented in an amorphous dispersion with PVP, nifedipine is able to diffuse far more quickly into the dissolution media.23
The first challenge in the development of our capsule formulation was to confirm the quality of the feed solution added to the capsule prior to freeze-drying. Using water – the traditional freeze-drying solvent – proved impossible, as even in the presence of PVP a sufficiently concentrated solution of nifedipine was impossible to achieve. Tert-butanol (TBA) proved successful – it dissolved enough of the drug to deliver a dose of 10mg of nifedipine to each capsule and the vapour pressure above its frozen solid allowed a good rate of sublimation. Thus, the feed solution contained just nifedipine, PVP and TBA. During development, solubility measurements of the nifedipine in this solvent system were required for each polymer drug ratio, grade and batch of the polymer.
Typical QC tests for freeze-dried amorphous cakes include quantification of residual water content, determination of the glass transition and collapse temperature, uniformity of weight and content. The additional QC challenges for our capsule concerned drug purity and capsule performance. Considering purity, nifedipine suffers from light-induced degradation, so chemical purity had to be confirmed during each stage of manufacture and also during testing. Simply excluding light from the feed solutions, freeze-drier and the dissolution testing, achieved specification. Hard gelatin capsules that contain a GRAS (generally recognised as safe) red colouring were used to maintain the drug content during storage (Table 1). The stability indicating HPLC assay developed for this study was able to identify the degradation products and showed that the amorphous formulations suffered significant chemical instability, but this could be arrested by the use of red capsules instead of the clear form that were initially used.
Table 1: Summary of stability study for the novel in-situ freeze-dried capsule formulation
All data = averages ± SE of n=6
Quality parameters | Quality control test | Specification | Average values ± standard error | |||||
0 months | 3 months 25 oC 57% RH | 3 months 37 oC 74% RH | ||||||
Physical stability | Weight of formulated capsule (mg) | 201 ± 20 | 201.57 | ± 0.3 | 200.4 | ± 0.3 | 201.06 | ± 0.7 |
Tg (oC) 2nd heat | – | 130.3 | ± 3.6 | 125.0 | ± 2.2 | 125.2 | ± 0.8 | |
Crystalline NIF % w/w | 0% | 0.00 | ± 0.0 | 0.0 | ± 0.0 | 0.0 | ± 0.0 | |
Chemical stability | Degradation products (%)* | <5% | 0.00 | ± 0.0 | 0.0 | ± 0.0 | 0.0 | ± 0.0 |
Performance of formulation | Drug content (mg) | 10 ± 0.5 | 9.6 | ± 0.12 | 9.4 | ± 0.1 | 9.4 | ± 0.1 |
Rate constant for dissolution, k, (min-1) | – | 0.4 | ± 0.1 | – | – | 0.4 | ± 0.0 | |
T80 (min) | < 20 min | 4.8 | ± 0.2 | – | – | 5.3 | ± 0.1 | |
* Degradation products include: 4-(2-nitrosophenyl)-pyridine homologue and 4-(2-nitrophenyl) pyridine homologue
QC testing supporting development
The early incorporation of pharmacopeia-recommended QC testing proved very important to the development of the new nifedipine capsule. For example, when investigating the optimum drug to polymer ratio, simply considering the BP uniformity of weight and drug content revealed that formulations with 10% w/w nifedipine in PVP complied more closely with BP regulations, while higher nifedipine to PVP content showed high variations (RSD% >2%) in unit dosage weight. The prototypes all contained the same 10mg dose of nifedipine, but with varied amounts of PVP; formulations containing < 50% w/w with respect to PVP had drug contents below the BP acceptable range of 10 ± 0.5 mg. It was concluded that low PVP content can reduce the strength of the FD cake, making it more susceptible to yield loss while under the high vacuum of the freeze-drying process, thus leading to variations in weight and the loss of drug. A simple conclusion, but derived from the early use QC testing.
A similar approach was used to discover the best composition to provide a fully amorphous formulation. Decreasing the ratio of nifedipine to PVP lowered the amount of crystalline nifedipine in the freeze-dried formulations. Using a differential scanning calorimetry, a quality-control-based assay was developed to quantify crystalline content in manufactured formulations. Capsules containing ≤50% w/w nifedipine in PVP showed an absence of crystalline nifedipine, a key specification to ensure enhanced dissolution rate. Furthermore, glass transition temperatures increased as the %w/w of nifedipine in PVP decreased.
Maintaining a high Tg is an important means of promoting physical stability of amorphous drug, thus it was set as a QC test for the novel in-situ freeze-dried capsule formulation to ensure desirable specifications are maintained (Table 1). In addition to the absence of crystallinity, the specifications set out in Table 1 include the QC dissolution test, which was based on the BP specification for the marketed fast-release nifedipine capsules and was set at 20 minutes for 80% dissolved.
QC consideration for the freeze-dried hard gelatin capsule shells
The effect of the freeze-drying process on the quality of gelatin hard capsules was also investigated following the World Health Organization (WHO) general monograph for quality control of capsules.
FT-IR analysis of treated and untreated capsules was compared to determine if the process of freeze-drying had caused chemical changes. The correlation between the two sets of capsules (for the range 4,000-400 cm-1) was >97%, indicating that no chemical changes to the gelatin capsule shell were induced by freeze-drying. Furthermore, following the WHO guidelines, the uniformity of mass of the treated gelatin hard capsules was determined. All 20 capsules tested showed less than 2% deviation from the average weight, therefore complying with the WHO limit of 10% deviation from the average weight.
Following the international pharmacopeia guidelines24 on disintegration testing of hard capsules, freeze-dried capsules were tested using the basket-rack apparatus described in section 5.3 of the international pharmacopoeia.24 All freeze-dried capsules disintegrated in 5.1 minutes, while the non-freeze-dried capsules disintegrated within 5.5 minutes. Thus, the freeze-drying process did not significantly affect the disintegration performance of the hard gelatin capsule shells. Both freeze-dried and non-freeze-dried capsules complied with the international pharmacopoeia limits for hard gelatin capsule disintegration time of <30 minutes.24
Considering and meeting the challenge of quality control early within an academic-led research project has proved to be extremely important, in showing that our new platform technology works, but also that it can meet, and in some cases outperform, the pharmacopeia specifications.
About the authors
 DR ABDULMALIK ALQURSHI is Lecturer in Pharmaceutics at Taibah University in Saudi Arabia, and Head of the Pharmaceutics and Pharmaceutical Technology Department. Dr Alqurshi’s research focuses on the design and development of novel oral formulations with enhanced disintegration and dissolution rates, as well as transferring bench formulations into Good Manufacturing Practice environments.
DR ABDULMALIK ALQURSHI is Lecturer in Pharmaceutics at Taibah University in Saudi Arabia, and Head of the Pharmaceutics and Pharmaceutical Technology Department. Dr Alqurshi’s research focuses on the design and development of novel oral formulations with enhanced disintegration and dissolution rates, as well as transferring bench formulations into Good Manufacturing Practice environments.
 DR PAUL G ROYALL is Lecturer in Pharmaceutics at King’s College London, and Programme Director of the MSc in Pharmaceutical Analysis and Quality Control. Dr Royall’s research focuses on the use of materials science for the development of new dosage forms, especially freeze-dried and amorphous formulations to be administered orally.
DR PAUL G ROYALL is Lecturer in Pharmaceutics at King’s College London, and Programme Director of the MSc in Pharmaceutical Analysis and Quality Control. Dr Royall’s research focuses on the use of materials science for the development of new dosage forms, especially freeze-dried and amorphous formulations to be administered orally.
References
- Kaur JG, Aggarwal GS, Rana AC. Improvement of drug solubility using solid dispersion. International Journal of Pharmacy and Pharmaceutical Sciences. 2012;4(2):47-53.
- Vasconcelos T, Sarmento B, Costa P. Solid dispersions as strategy to improve oral bioavailability of poor water soluble drugs. Drug Discovery Today. 2007;12(23-24):1068-1075.
- Streubel A, Siepmann J, Bodmeier R. Drug delivery to the upper small intestine window using gastroretentive technologies. Curr Opin Pharmacol. 2006;6(5):501-8.
- Gardner DR, Casper R, Leith F, and Wilding I. Noninvasive methodology for assessing regional drug absorption from the gastrointestinal tract. Pharmaceutical Technology International. 1997;9(Jun):46,48,50,52-53.
- Thomas VH, Bhattachar S, Hitchingham L, Zocharski P, Naath M, Surendran N, Stoner CL, El-Kattan A. The road map to oral bioavailability: an industrial perspective. Expert Opinion on Drug Metabolism & Toxicology. 2006;2(4):591-608.
- Leuner C, Dressman J. Improving drug solubility for oral delivery using solid dispersions. Eur J Pharm Biopharm. 2000;50(1):47-60.
- Kumar A, Sahoo SK, Padhee K, Kochar PPS, Satapathy A, Pathak N. Review on solubility enhancement techniques for hydrophobic drugs. Pharm. Globale. 2011; 2(Copyright 2013 American Chemical Society (ACS). All Rights Reserved.): No pp. given.
- Chaumeil JC. Micronization: A method of improving the bioavailability of poorly soluble drugs. Methods and Findings in Experimental and Clinical Pharmacology. 1998;20(3):211-215.
- Vogt M, Kunath K, and Dressman JB. Dissolution enhancement of fenofibrate by micronization, cogrinding and spray-drying: Comparison with commercial preparations. European Journal of Pharmaceutics and Biopharmaceutics. 2008;68(2):283-288.
- Ali SL, Nifedipine. Anal. Profiles Drug Subst. 1989; 18(Copyright 2013 American Chemical Society (ACS). All Rights Reserved.):221-88.
- Kaushal AM, Gupta P, Bansal AK. Amorphous drug delivery systems: molecular aspects, design, and performance. Crit Rev Ther Drug Carrier Syst. 2004;21(3):133-93.
- Craig DQM, Royall PG, Kett VL, Hopton ML. The relevance of the amorphous state to pharmaceutical dosage forms: glassy drugs and freeze dried systems. International Journal of Pharmaceutics. 1999;179(2):179-207.
- Trasi NS, Baird JA, Kestur US, Taylor LS. Factors Influencing Crystal Growth Rates from Undercooled Liquids of Pharmaceutical Compounds. J. Phys. Chem. B. 2014;118(33):9974-9982.
- Baird JA, Van Eerdenbrugh B, Taylor LS. A classification system to assess the crystallization tendency of organic molecules from undercooled melts. J. Pharm. Sci. 2010; 99(9):3787-3806.
- Yu L. Amorphous pharmaceutical solids: preparation, characterization and stabilization. Advanced drug delivery reviews. 2001;48(1):27-42.
- Bhugra C, Pikal MJ. Role of thermodynamic, molecular, and kinetic factors in crystallization from the amorphous state. Journal of pharmaceutical sciences. 2008;97(4):1329-1349.
- Shah B, Kakumanu VK, Bansal AK. Analytical techniques for quantification of amorphous/ crystalline phases in pharmaceutical solids. Journal of Pharmaceutical Sciences. 2006; 95(8):1641-1665.
- Tropin TV, Schmelzer JWP, Schick C. On the dependence of the properties of glasses on cooling and heating rates I. Entropy, entropy production, and glass transition temperature. Journal of Non-Crystalline Solids. 2011;357(4):1291-1302.
- Kerc J, Srcic S. Thermal-analysis of glassy pharmaceuticals. Thermochimica Acta. 1995;248:81-95.
- Vessot S, Andrieu J. A Review on Freeze Drying of Drugs with tert-Butanol (TBA) + Water Systems: Characteristics, Advantages, Drawbacks. Drying Technol., 2012. 30(Copyright 2013 American Chemical Society (ACS). All Rights Reserved.):377-385.
- Courtney P, Alqurshi A, Forbes B, Strang J, Royall PG. Freeze-dried formulations: a new perspective on reformulating naloxone. European Pharmaceutical Review. 2017;22(3):32-36.
- Alqurshi A, Kumar Z, McDonald R, Strang J, Buanz A, Ahmed S, Allen E, Cameron P, Rickard JA, Sandhu V. Amorphous formulation and in vitro performance testing of instantly disintegrating buccal tablets for the emergency delivery of naloxone. Molecular pharmaceutics. 2016;13(5):1688-1698.
- Alqurshi A, Chan KA, Royall PG. In-situ freeze-drying-forming amorphous solids directly within capsules: An investigation of dissolution enhancement for a poorly soluble drug. Scientific Reports. 2017;7.
- The International Pharmacopoeia Fifth Edition. 10-09-2015. WHO.