

Is high resolution mass spectrometry the missing piece in continuous bioproduction?
Posted: 5 August 2017 | Jonathan Bones, Michael Blank, Rowan Moore, Simon Cubbon | No comments yet
Consistent with quality-by-design (QbD), and driven by the desire for continuous manufacture and real-time lot release, biopharmaceutical manufacturers require advanced process analytical technologies (PAT) for process monitoring and rapid analytical information generation.
In addition to the online monitoring of cell culture pH, temperature and dissolved oxygen, the complexity of therapeutic proteins dictates that technologies delivering near real-time full structural insights into the drug substance are also needed.
Several time-consuming, intensive critical quality attribute (CQA) characterisation methods are performed offline during bioproduction. The drive towards continuous, smaller-scale, single-use perfusion based bioprocessing technologies means that culture durations will be extended, necessitating methods that are able to rapidly monitor multiple CQAs in an online format. Ultra-high-performance liquid chromatography (UHPLC) and mass spectrometry (MS) are already successfully used at-line/offline during bioproduction and for lot release. High-resolution LC-MS is growing in popularity, based on its ability to efficiently offer a wealth of multi-attribute information that can be used to optimise manufacturing processes and improve drug safety and efficacy.
Barriers to the adoption of LC-MS in an online environment will be discussed along with recent advances, including the potential application of CGMP-compliant MS characterisation throughout bioprocessing and lot release.
PAT and biopharmaceutical manufacturing
PAT has been defined by the United States Food and Drug Administration (FDA) as: “A mechanism to design, analyse, and control pharmaceutical manufacturing processes through the measurement of critical process parameters (CPP) which affect CQAs.”1
As a result of concerted efforts over the last decade, PAT implementation in the manufacture of small molecule pharmaceuticals is growing. This effort was driven by:2
- The forecasted benefits of moving from batch-based manufacturing to continuous manufacturing
- The subsequent ability to manufacture multiple active pharmaceutical ingredients (APIs) and be more responsive to changing market demand
- ‘Real-time-release’ (RTR), smarter manufacturing processes with feed-back and feed-forwards mechanisms
- The promise of a leaner manufacturing outfit
- Significant economic benefits.
For the biopharmaceutical industry, these benefits are yet to be realised at the production-scale, with PAT and online sampling/real-time analysis still being a largely research-based exercise. A transition to single-use biotechnologies, continuous bioprocessing and the implementation of PAT could afford up to 55% cost savings over a 10-year period.3,4 Considering the higher costs associated with biopharmaceutical production, such savings would be significant.
Since the same technical and business drivers exist for the development of on-line, fully integrated multi-attribute monitoring (MAM) for the characterisation of expressed therapeutic proteins in near real time, why is it taking biopharma so long to catch up?
Challenges of implementing PAT
Several unique challenges face the analysts and process/scale-up engineers implementing PAT in a bioproduction environment:
- Matrix complexity
- Analyte concentration (relatively low)
- Technological limitations/selectivity
- Fluidic movement
- Maintaining sterility
- Fouling of probes/sampling devices
- Obtaining cell-free samples.
Online measurements are already recorded in the modern bioproduction facility, either via external monitoring (off gas/environmental), off/at-line sample analyses (extracting and taking the sample to the lab) or, more recently, via in-bioreactor mobile sensing devices.5 These measurements provide analysts and engineers with a near real-time update on the environment within the bioreactor, cell and media behaviour/state, and the titre and attributes of the drug substance. For biologic drug products, which are larger and more complex than their small molecule counterparts, more informative technologies than simple spectroscopic (eg, Raman) and biochemical tests (eg, ELISA) are required.
Analytical testing of in-process samples from bioreactors and purified drug substance is performed off-line in QC laboratories using several individual, single-quality, attribute-focused methods that require significant sample transport, processing, analysis and data interpretation. These methods typically require multiple analyses to be performed in parallel, and are often dependent upon comparison to a reference sample, using pattern matching rather than providing the type of in-depth structural characterisation that can be obtained via LC-MS approaches. Furthermore, post-translational modification (PTM) induction is also possible within the chain of custody, from sample collection to final analysis. The requirement for analytical testing and associated sample purification steps delays data turnaround and the ability to process data from many bioreactors in parallel, which is often the case during process development experimental designs.
The ongoing transition in bioproduction from steel bioreactors to single-use bioprocessing technologies and the move from batch to continuous manufacturing are compounding existing analytical challenges. This (potentially) adds another dimension to PAT, involving risk assessment and mitigation in the study, understanding and control of potential extractable and leachable impurities.6
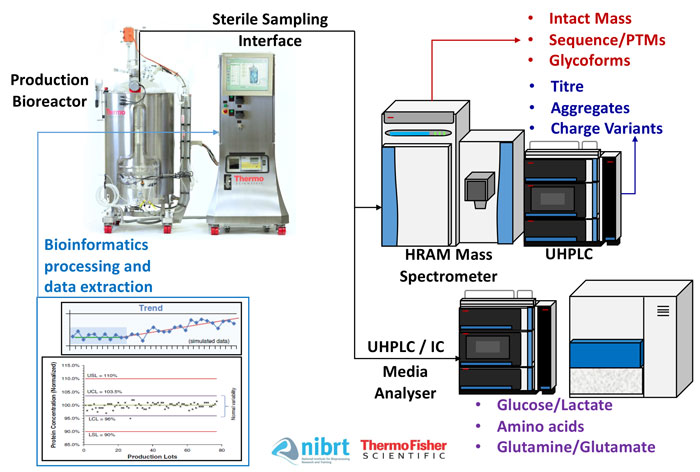
Figure 1: Bring the lab to the sample: an overview of a multi-attribute analytical platform
Opportunities and potential solutions
The solution may lie, perhaps unexpectedly, in the perceptually complex world of UHPLC and high-resolution accurate-mass (HRAM) spectroscopy. Online UHPLC-HRAM permits the analysis of multiple CQAs on a single analytical platform. Coupling such a system with online aseptic sampling, incorporated fluidics and integrated informatics allows the lab to be brought to the sample, rather than the sample to the lab, greatly reducing the turnaround time for actionable knowledge and an ultimately higher quality, more cost-effective bioproduction strategy. This type of platform can perform analytical workflows such as intact protein analysis (titre, aggregates, charge variants, intact mass – either denatured or native), middle-up/down analyses, peptide mapping (post-translational modifications and sequence verification), released glycan analysis and/or glycopeptide analysis.7
Automatically sampling, manipulating and analysing bioreactor samples using an ‘end-to-end multi-attribute analytical solution’ can generate a detailed product quality (PQ) profile much more quickly, potentially in under six hours (Figure 1). This presents the opportunity for a uniquely innovative analytical platform that can enable enhanced agile process characterisation and better process control strategies. Bioinformatics and associated software developments would enable a simplified visualisation interface that could interpret the multi-attribute analytical data.
Mass spectrometry in drug development
MS, specifically systems delivering HRAM, are commonly used in R&D. HRAM spectroscopy coupled to UHPLC provides significant insights into complex biologics, from high-level intact mass analysis under native or denaturing conditions, or sub-unit mass analysis, through to detailed peptide mapping. Indeed, these complex biologics would not reach clinical trials if it were not for the data produced during HRAM characterisation studies.
When drug development passes through into chemistry, manufacturing and controls (CMC), which encompasses early/late development, scale up and pilot plants, through to manufacturing and QC/lot release, MS (particularly HRAM MS) is the exception rather than the rule. There are several reasons for this:
- Historically, HRAM MS required skilled users to operate the systems
- Processing has been complex to learn, slow to process and lacking compliance readiness
- Traditional QC and lot-release methodologies are well known and understood
- Risks and cost of implementation of new technologies.
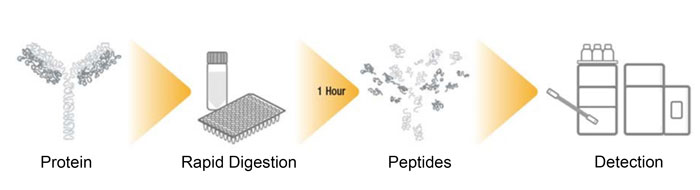
Overview of MAM workflow
With regulatory agencies such as the FDA and EMA shifting towards a QbD approach, as outlined by International Conference on Harmonisation (ICH) guidelines Q8-11,8-11 the market must evaluate and adopt new technologies. Advances in manufacturing and process analytics should yield longer term savings from increased efficiencies, improved product knowledge, the potential for shifting towards continuous manufacturing, and because lot-release testing is no longer required (as the product is continually monitored).
QbD guidelines recommend developing a quality target product profile (QTPP),12 with CQAs identified and an overall strategy for maintaining QTPP outlined. This begins before therapeutics can be used within clinical trials and engages numerous different separation and detection techniques to characterise these therapeutics in painstaking detail. Traditional characterisation includes a diverse battery of techniques, ranging from size-exclusion, hydrophobic-interaction, ion-exchange and reversed-phase chromatography, to capillary electrophoresis (reduced or non-reduced) sodium dodecyl sulfate gels, and ELISA, to name but a few.
These methods can be lengthy, and while this is less of an issue early on, when rapid and high throughput is required (eg, when moving into CMC) these methods place a burden on the analysts to deliver rapid but nevertheless accurate results. This is where the use of HRAM spectrometry for MAM can offer significant improvements:
- One single experiment can replace several conventional methods
- Multiple attributes can be monitored within this single method
- Greater confidence and accuracy can be obtained, as well as additional information not accessible using traditional methods
- Significant time savings and increased throughput can be realised.
Table 1: Overview of conventional release methods that can be covered by peptide mapping MAM
| Conventional Release Methods | ||||||
Critical Quality Attribute Assessment | SEC | CEX | rCE‐ SDS | nrCE‐ SDS | HILIC | ID ELISA | HCP ELISA |
CDR Tryptophan Degradation | Indirect | – | – | – | – | – | – |
C‐terminal Amidation | – | Indirect | – | – | – | – | – |
C‐terminal Lysine | – | ✓ | – | – | – | – | – |
Deamidation | – | Indirect | – | – | – | – | – |
Glycation | – | – | ✓ | ✓ | – | – | – |
HCP | – | – | – | – | – | – | ✓ |
High Mannose | – | – | – | – | ✓ | – | – |
Hydroxylysine | – | – | – | – | – | – | – |
Identity | – | ✓ | – | – | – | ✓ | – |
N‐glycosylation | – | – | ? | ? | – | – | – |
N‐terminal pyroGlutamate | – | Indirect | – | – | – | – | – |
Unusual Glycosylation |
| Indirect | ? | ? | ✓ | – | – |
Multi-attribute monitoring (MAM)
Using MS within the QC environment is not necessarily a novel concept when it comes to single measurements, or when a single characteristic is being monitored, but the application of HRAM spectrometry to provide as much information as possible within a single analysis is very exciting. The concept of MAM is that, within a single experiment, numerous attributes (which would typically be determined by several separate analyses) are monitored using a confirmatory analysis, where ‘gold standard’ reference data is compared to multiple samples in terms of differential CQA profile and corresponding critical process parameters (CPPs), allowing decisions to be made in near-real time. MAM is today realised through a peptide mapping workflow (Figure 2), where the biotherapeutic is rapidly digested using ‘soft’ conditions to minimise process-induced modifications. Peptides are separated and detected using UHPLC coupled to HRAM MS and finally processed using compliant software, typically using integrated informatics such as a chromatography data system (CDS) connected to a laboratory information management system (LIMS), which would be commonplace within this environment. This MAM workflow will have been carefully optimised to provide absolute confidence through highly reproducible digestions, separations and superlative mass accuracy, all with throughput in mind. Table 1 highlights conventional release methods and the attribute assessment that they can deliver. All these methods and attributes can be measured in a single peptide mapping-based MAM experiment. Over 10 further CQAs can be monitored as part of the MAM experiment, so time to result can be significantly reduced while increasing product knowledge and understanding. While MAM cannot replace all the conventional release methods – because it cannot measure aggregation, for example – it is a viable option for rapidly speeding up access to critical information throughout the CMC process. With HRAM MS being adopted within this arena it also opens up the opportunity to hyphenate MS with more conventional chromatographic methods, such as SEC, to provide native intact mass confirmation.
Conclusions
The availability of MAM, based on UHPLC and HRAM MS, will drive process development and optimisation studies, and enable complete product and process characterisation, with data returned in near real time allowing for enhanced levels of bioprocess control. Complete systems consisting of automated sampling and purification with online bioreactor profiling, product quality analytics and a simplified graphical user interface comprising of trend and quality plots for simplified visualisation of the product and process performance data, are desired.
MAM will become an essential system for future process development, intensification and control during batch and continuous manufacture of biopharmaceuticals, offering the potential for end users to gain an increased understanding of their manufacturing processes and therapeutic products with significant potential for associated new intellectual property concerning both to be discovered.
Perhaps ‘MS everywhere’ really is on the horizon – upstream, downstream, drug formulation, fill and finish.
About the authors
 ROWAN MOORE, PhD, is Pharma and Biopharma Applications Manager, HPLC, IC and Chromatography Consumables, at Thermo Fisher Scientific. Dr Moore specialises in the development of advanced chromatographic and mass spectrometric analytical solutions for the bio/pharmaceutical industry. Since gaining her PhD in Veterinary Preclinical Science, focused on proteomics and metabolite identification, from the University of Liverpool in 2008, Dr Moore has worked in high-throughput bioanalysis and for leading instrument manufacturers delivering LC and HRAM MS workflows and solutions.
ROWAN MOORE, PhD, is Pharma and Biopharma Applications Manager, HPLC, IC and Chromatography Consumables, at Thermo Fisher Scientific. Dr Moore specialises in the development of advanced chromatographic and mass spectrometric analytical solutions for the bio/pharmaceutical industry. Since gaining her PhD in Veterinary Preclinical Science, focused on proteomics and metabolite identification, from the University of Liverpool in 2008, Dr Moore has worked in high-throughput bioanalysis and for leading instrument manufacturers delivering LC and HRAM MS workflows and solutions.
 SIMON CUBBON, PhD, is Pharma and Biopharma Applications Manager, Informatics, at Thermo Fisher Scientific. Dr Cubbon has worked within the analytical instruments industry for over 10 years, holding a variety of positions from applications scientist through to informatics product management. He studied for his PhD at the University of York, where he was interested in using LC-MS to study the metabolic profile of long bone fractures.
SIMON CUBBON, PhD, is Pharma and Biopharma Applications Manager, Informatics, at Thermo Fisher Scientific. Dr Cubbon has worked within the analytical instruments industry for over 10 years, holding a variety of positions from applications scientist through to informatics product management. He studied for his PhD at the University of York, where he was interested in using LC-MS to study the metabolic profile of long bone fractures.
 JONATHAN BONES, PhD, has been Principal Investigator, NIBRT Characterisation and Comparability Laboratory, since 2012. Dr Bones’ research has focused on the development and application of advanced LC-MS and CE-MS solutions to solve challenges associated with the characterisation and comparability assessment of therapeutic proteins and manufacturing processes. He received his PhD in Analytical Chemistry from Dublin City University in 2007.
JONATHAN BONES, PhD, has been Principal Investigator, NIBRT Characterisation and Comparability Laboratory, since 2012. Dr Bones’ research has focused on the development and application of advanced LC-MS and CE-MS solutions to solve challenges associated with the characterisation and comparability assessment of therapeutic proteins and manufacturing processes. He received his PhD in Analytical Chemistry from Dublin City University in 2007.
 MICHAEL BLANK, PhD, is Product Marketing Manager at Thermo Fisher Scientific. Dr Blank specialises in mass spectrometry-based techniques for quality control, product characterisation and lot release, as well as numerous peptide quantitation techniques. Michael received his PhD in Chemistry from Stanford University in 2012.
MICHAEL BLANK, PhD, is Product Marketing Manager at Thermo Fisher Scientific. Dr Blank specialises in mass spectrometry-based techniques for quality control, product characterisation and lot release, as well as numerous peptide quantitation techniques. Michael received his PhD in Chemistry from Stanford University in 2012.
References
To view references, please visit: www.europeanpharmaceuticalreview.com/4-17-Moore
Issue
Related topics
Related organisations
The National Institute for Bioprocessing Research and Training (NIBRT), Thermo Fisher Scientific





