

Endotoxin masking hold-time study performance parameters
Posted: 15 April 2017 | Kevin L Williams (Hyglos GmbH a BioMerieux company) | No comments yet
Presented here are recommendations for performing an endotoxin masking hold-time study. The US Food and Drug Administration (FDA) and the European Medicines Agency (EMA) have requested hold-time studies to determine the presence of what has been called ‘low endotoxin recovery’ (LER) for submission with biologic licence applications (BLA)…

Performance parameters to be discussed include:
- Drug formulations
- Spike type and amount
- Timing and temperature of hold
- Methodology
- Container effects
- Test kinetics
- Test interpretation
- Implications
- An example protocol.
The parameters discussed constitute a recommended initial screen, which can be made more specific to match specific process parameters if masking is determined to occur.
1. Formulation
Regulators charged with approving BLAs have defined LER-subjected formulations as those containing polysorbate (20 or 80). Typically, formulations also contain a chelator such as a citrate or phosphate buffer and, in some cases, histidine has been implicated. Some companies have included biologic therapeutic protein solutions that do not contain polysorbate in hold time studies because the masking effect, regardless of the mechanism, can obscure the ability to determine endotoxin either as bound to protein or as per the specific LER mechanism. Protein binding has been known for decades, yet the systematic exclusion of its presence in analytical testing seems neglected. From the Petsch et al studies1 performed 20 years ago, in which researchers used model proteins including IgG (the prototypical structure for monoclonal antibodies) and showed that recovery of endotoxin in low concentrations (1mg/ml) of protein in pH neutral solutions (pH7) gave almost complete binding of endotoxin by the protein (<2% recovery by LAL test). After protease treatment, the recovery was around 90%. While protease treatments are difficult and often commercial proteases are contaminated with low levels of endotoxin, the demasking scheme developed by Hyglos has been shown to work for protein-bound endotoxin detection as well as for LER solutions.
2. Spike
The endotoxin type used for spiking has been subject to debate by industry participants, with some maintaining that LER only occurs with purified lipopolysaccharide. However, Hyglos has shown repeatedly that various ‘naturally occurring endotoxins’ (NOE) are masked under LER conditions and is now preparing a definitive answer to the question in further studies. The choice of reference standard endotoxin (RSE) gives the user an advantage in that different LAL types can be explored relatively easily should the need arise. One can also choose control standard endotoxin (CSE) in the desire to stick to a routinely preferred LAL.
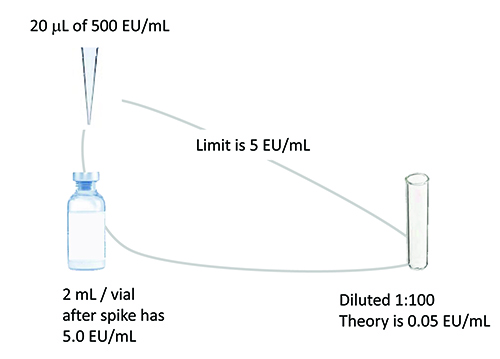
Figure 1: Example spike for hold time study relates amount of spike to product
volume / limit to dilution needed to overcome interference.
The spike volume should be minimised to avoid changing the concentration of the undiluted drug product. Typically, a 10 microlitre spike of a stock solution 100 times the endotoxin limit concentration per 1ml of product will suffice. The spike concentration chosen should be close to the drug limit concentration and at least 10 times higher than the detection limit after dilution. See Figure 1 for an example product spike. LAL reagent water controls should accompany initial spikes to serve as the baseline for spike recovery. A masking control is recommended to ensure that masking conditions have been met.
3. Timing and temperature
Since the initial screen surveys generic conditions, the recommendation to use room temperature masking and an interval out to 14 days is based on regulatory observations. For temperature, a manufacturing process step is rarely exclusively refrigerated. Any process has a beginning time and an ending time (such as product filling), in which the product sits unrefrigerated. Time at room temperature has a profound effect on masking protein solutions. Similarly, in choosing a short masking study (seven days), one may later learn that sometimes the process can be extended. One does not want to repeat such studies due to an initial estimation that later proves to be a borderline assumption.
4. Methodology
The test is performed by either the straightforward ‘chronological’ method, which is performed by spiking and testing samples as time points arise, or by the ‘reverse’ spike method which seeks to reduce test variability by ensuring that all samples end up on the same test plate at time zero. An example protocol is included below for such a reverse study.
5. Container effects
If using the original container, as has been recommended by regulatory agencies, container effects are automatically accounted for in the masking study. In some cases, it may be difficult to use the original container, including vials or syringes with a lack of headspace or the lack of an adequate opening to allow pipetting of sample in and out. However, if using a secondary container, one should be aware of differences in recovery that can occur due to glass. Some pretesting may be useful to ensure that the study is about the drug product and not the container itself.
6. Test kinetics
Test masking kinetics is an examination of the recovery values over time. A decline after an initial adequate recovery is typical for LER studies, and the rapidity of the decline indicates the masking capability of the solution. One can feel more confident of the effect when the kinetics are occurring within the first couple of days or even the first couple of hours after spike. Figure 2 shows typical kinetic graphs for recovery over time of spike into undiluted product. Note that both the LER-subjected endotoxin as well as the protein-bound endotoxin quickly disappear, but the reduction over time may level off with protein binding, whereas the classical LER mechanism will go to completion.
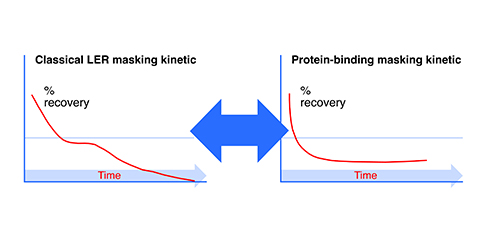
Figure 2: Endotoxin masking kinetics.
7. Test interpretation
LER is just what it says: low endotoxin recovery. ‘Low’ is defined as less than 50% for two consecutive time point recoveries over the course of the study. Sample spike recoveries can be compared to either theoretical expectations or recovery of water control spikes. Theoretical recoveries should be used only when water control recoveries are within the acceptable range.
8. Implications
The implications of endotoxin masking are straightforward in that LAL testing of masked samples gives little assurance of detection of endotoxin arising from potential contamination events. Biologics have a life-saving history, but also one marred by sporadic and tragic immunogenicity events.2 The most recent philosophy of contamination control has sought to minimise two critical elements, both of which constitute the two arms of the adjuvant effect:
- Aggregates, particulates and emulsions (including leachables and extractables) that the body can perceive as (mimicking) infectious agents. Here drug manufacturers have made enormous strides after costly initial efforts.
- Microbial-derived molecules3 including endotoxin that activate toll-like receptors.
Even endotoxin considered non-fever-causing is immunogenic, as seen in the use of a detoxified, non-pyrogenic endotoxin as a vaccine adjuvant as well as the other toll-like receptor (TLR) activators as per recent FDA research.4,5,6 Therefore detecting endotoxin in its various molecular forms is important in biologics in a way that it was not in small molecule drugs.
Detection based solely on fever, as a systemic response, is past its prime in providing adequate assurance of safety in this important class of drugs. The prevalence of fever in many biologics may exclusively arise from their mode of action in modulating the immune system, but until a more complete control of toll-like receptor activating microbial impurities is gained (as increased control has only recently been gained for particulates and aggregates), there cannot be complete confidence in this. Two points to consider in this regard:
- Human therapeutic proteins (as opposed to chimeric or animal derived) should not be immunogenic, as even microbial (sub-unit) proteins need an adjuvant to produce adequate immune responses in vaccinations.7,8
- The adjuvant effect is a proven cause of immunogenicity in terms of protein aggregation, and particulate occurrence and has been thoroughly accepted in terms of lot-to-lot exclusion, whereas the other arm of the adjuvant effect: microbial-derived molecules, has not been widely accepted (even by USP9) and therefore has, to date, not received a similar rigorous effort at exclusion. USP maintains that ‘no fever’ and ‘no ADRs’ occur due to endotoxin in biologics.10
Three things make the USP stance problematic:
- Fever is common in biologics administration11
- Fever is not considered an ADR where the expectation of fever is written into the package insert (as derived from clinical trial data)12
- Fever from biologic drug mode of action is indistinguishable from fever from endotoxin administration.13
9. Example protocol
See Figure 3 for an overview of samples, controls and standards to be analysed.
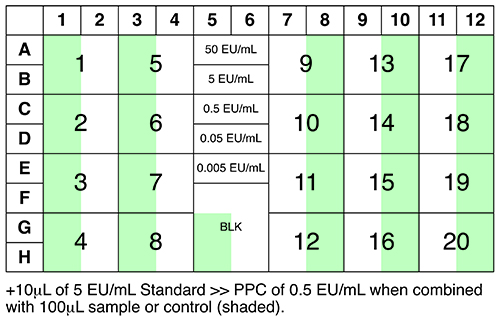
Figure 3: Suggested plate layout.
Summary
Hold-time studies are an important part of an overall microbiological control strategy for biologic drug manufacture. Importantly, the context for control has changed with the introduction of immunological concerns due to adverse drug events associated with biologic drug administration. For some, business as usual remains the message du jour, however, as seen in the recent application of stricter standards governing aggregates and particulates, one can expect to see a renewed focus on microbial-derived artifacts that include the innate immune response modulating impurity (IIRMI) or adjuvant-based perspective. After all, this perspective can simply be viewed as the ‘contaminant effect’, in that anything existing in a protein therapeutic that is not found in the natural protein in its natural environment (human blood) can be viewed by the immune system as a contaminant via pattern recognition receptors.
This fairly simple idea of control has governed all the efforts to date to reduce immunogenicity (see Figure 4), including:
- Replacement of foreign sequences in drug molecule design (humanisation)
- Prevention of protein aggregation in manufacturing performance
- Preclusion of particulates and emulsions in all aspects of production
- Extending to product containers and closures (including leachables and extractables such as coatings like silicone, etc).
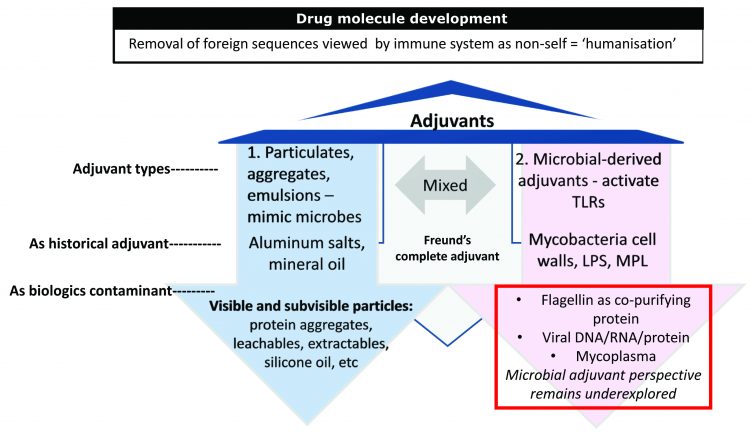
Figure 4: Known causes of immunogenicity. The two arms of the adjuvant effect can be viewed in the historic use of adjuvants in the vaccine world: (1) particulates including aluminum salts and oil in water emulsions as added to vaccine proteins and (2) microbial-derived adjuvants including mycobacterium cell walls (Freund’s complete adjuvant) and LPS (monophosphoryl lipid A) added to vaccine proteins to bolster immune responses (immunogenicity). Immunogenicity is necessary in vaccines but feared in biologics.
There can be little doubt that microbial-derived adjuvants will be increasingly added to this list as testing can be devised.
References
- Petsch D. Proteinase K digestion of proteins improves detection of bacterial endotoxins by the Limulus amebocyte lysate assay: application for endotoxin removal from cationic proteins. Anal Biochem.1998, May 15;259(1):42-7.
- Kotarek et al. Consider Omontys pulled from the U.S. market after less than a year: subvisible particle content, formulation and dose of an erythropoietin peptide mimetic product are associated with severe adverse postmarketing events. Journ Pharm Sciences. 2016;105:1023-1027.
- MAMPs – microbial associated molecular patterns
- Casella CR, Mitchell TC. Putting endotoxin to work for us: monophosphoryl lipid A as a safe and effective vaccine adjuvant. Cell Mol Life Sci.2008 Oct;65(20):3231-40. doi: 10.1007/s00018-008-8228-6.
- Verthelyi D, Wang V. Trace Levels of Innate Immune Response Modulating Impurities (IIRMIs) Synergize to Break Tolerance to Therapeutic Proteins, FDA. 2010. http://journals.plos.org/plosone/article?id=10.1371/journal.pone.0015252
- Haile LA, Puig M, Kelley-Barker L, Verthelyi D. Detection of Innate Immune Response Modulating Impurities in Therapeutic Proteins, 2015. http://journals.plos.org/plosone/article?id=10.1371/journal.pone.0125078
- But then again, USP did not accept higher particle constraints until after the Omontys deaths. Now there is USP <787> in addition to USP <788>. Carpenter et al. Overlooking subvisible particles in therapeutic protein products: gaps that may compromise product quality. J Pharm Sci. Apr 2009;98: 4:1201-1205. This paper predicted the Omontys tragedy years earlier: “Thus, unless proper quality controls are in place for subvisible particles, a product that was safe and effective in clinical trials may unexpectedly cause adverse events in patients after commercialization.”
- “Infusion of many biologics, particularly mAbs, provokes a characteristic infusion syndrome, usually within one or a few hours during/after the first administration. Whereas most reactions are mild to moderate with symptoms often described as ‘flu-like’ with fever, chills, rigors, headache, nausea, asthenia, rash, and pruritus, a small number of patients, mostly at the first or second infusion, show potentially fatal symptoms resembling an IgE antibody-mediated reaction with hypotension, cardiac arrest, bronchospasm, and urticaria.” From: Safety of Biologics Therapy. Brian Baldo. Springer 2016.
- Manufacturer reported postmarketing events (CFR 314.80): “within 15 days of receipt … required to report to FDA individual case safety reports for events classified as ‘unexpected’ or ‘unlabeled’ (not described in the product package insert) and ‘serious’. Fever is not considered an ADR… Frassetto L, Tsourounis C. Improving the reporting of ADR in the hospital setting. Postgraduate Medicine. Nov 2010.
- “Endotoxin doses of 2-4 ng/kg body weight cause flu-like symptoms (fever, chills, myalgia (muscle pain), headache, nausea) and increases in blood TNF and IL-6 levels similar to what is seen in sepsis…” From: DellaGioia N, Hannestad J. A critical review of human endotoxin administration as an experimental paradigm of depression. Neurosci Biobehav Rev. 2010 Jan;34(1):130–143.





