

Identification of peptides and proteins in suspected illegal medicinal products using MALDI-TOF-MS
Posted: 26 February 2019 | Ahmad Amini, Henrik Lodén, Torgny Rundlöf | 1 comment
Matrix-assisted laser desorption/ionisation (MALDI) time-of-flight (TOF) mass spectrometry (MS) has proven to be an excellent technique for identification of illegally distributed peptides and proteins for human use. The identification of proteins is mainly based on peptide mass fingerprinting (PMF) – ie, the bottom-up approach – while the small proteins and peptides can be identified through the top-down approach. This paper reports on the application of MALDI for the identification of human growth hormone, melanotan II and delta sleep-inducing peptide…
Since 1992, with the coming of sensitive commercial instrumentation based on MALDI-TOF-MS, the technology has widely been used for protein and peptide identification.1,2 Since its development, MALDI-TOF-MS has been the primary instrumentation used for PMF,3,4 due to its high sensitivity and mass accuracy, speed, absence of multiple charge mass signals and relatively high tolerance toward additives and contaminants such as salts, matrix components and excipients.5 MALDI analysis requires only minimal sample treatment; ie, a few microlitres of sample and matrix are mixed and spotted on the MALDI target plate, which can also be automated. Furthermore, MALDI is a micro-destructive analytical technique and the remaining material on the MALDI target plate can be archived for later analysis. The high sensitivity of MALDI implies that only a small aliquot of a peptide or a protein digest is required, and the remainder can be used for alternative complementary measurements by other analytical techniques. Besides the accurate mass determination of peptides, MALDI provides additional information on the primary structure of the peptide by sequencing the peptide ions in post-source decay (PSD) mode.6 The predominant detection of singly charged peptide molecules by MALDI-MS facilitates the evaluation of PMFs significantly. In addition, MALDI provides sub-picomole limits of detection and a mass range in excess of 100kDa.
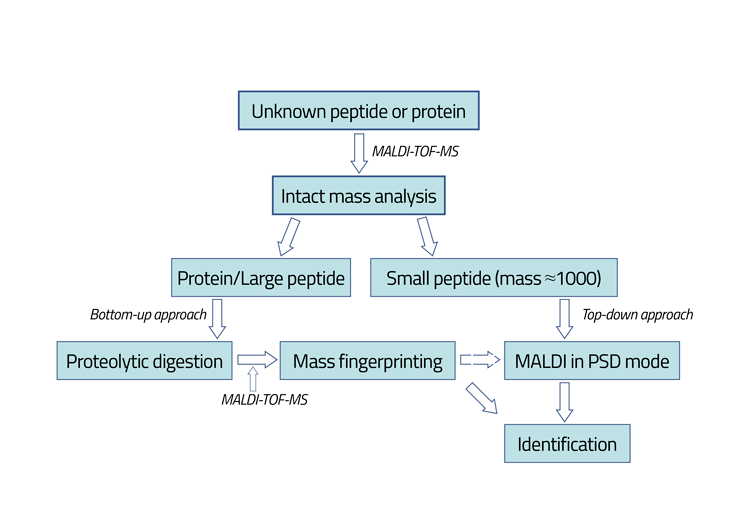
Figure 1: Schematic presentation of the protein/peptide identification by MALDI-TOF-MS. The sample being analysed is dissolved in 50mM ammonium bicarbonate (pH=7.5). One microlitre of the sample is then mixed with the matrix solution. The matrices being used for the analysis of native somatropin and the tryptic peptides have been sinapinic acid and a-cyano-4- hydroxycinnamic acid, respectively. Enzymatic degradation of analytes was performed by addition of trypsin into the analyte sample.
The concept of a proteolytic peptide pattern, ie, protein peptide mapping (PPM), being characteristic of a protein was first demonstrated by SDS-PAGE.7 In 1989, peptide sequencing by automated Edman degradation had a cycle time of nearly one hour per amino acid residue. Samples of interest often contained complex mixtures of proteins, which usually required separation by SDS-PAGE followed by electro blotting onto a PVDF membrane.8
However, a more rapid approach to protein identification is PMF. Using the bottom-up approach for PMF, the digested protein is analysed by MALDI-TOF-MS to generate mass-to-charge ratio (m/z) values in the mass spectrum, which in turn give rise to a characteristic ‘peptide mass fingerprint’ of the protein at nanogram level.9 The fingerprint serves to identify the protein through comparison with in silico digest of the corresponding reference standard.10 Trypsin is a commonly used proteolytic enzyme for PMF, since it is relatively cheap, highly selective and generates peptides with an average size of about 8–10 amino acids, which are ideally suited for analysis by MS. It cleaves principally on the C-terminal side of arginine (Arg) and lysine (Lys), with the exception of Arg-Pro and Lys-Pro.11 The drawback is that small peptides do not generate a peptide map that could be used for the identification. In such cases, the top-down approach is applied, ie, the intact peptide is subjected to PSD analysis in order to elucidate its amino acid sequence.
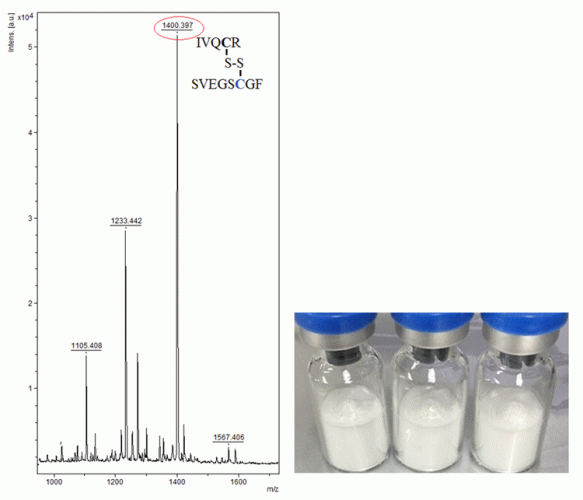
Figure 2: Peptide mass fingerprinting of human growth hormone, by MALDI-TOF-MS. The signature peptide characteristic for somatropin is marked in the spectrum. The photo below shows typical illegally distributed somatropin samples.
The singly-charged ions generated by MALDI-TOF-MS are a mixture of b-, y- and a-ions accompanied by ions resulting from neutral loss of ammonia or water.12 Protein identification, in the absence of a reference standard, is accomplished by using the PMF to search a protein sequence database by means of different search engines such as ProFound,13 MASCOT14 or SEQUEST.15 A value-based scoring system has been proposed that facilitates PMF-based protein identification without accompanying amino acid data.16 Those parameters considered to be important for the identification include: the isoelectric point (pI), molecular mass, protein sequence coverage and the number of matching proteolytic peptides.16 A scoring system has been developed where peptides are identified on the basis of identification points (IP).17 The molecular masses of both intact peptides and fragment ions are involved in the scoring system. The PMF-approach is based on bottom-up sequencing MS.10
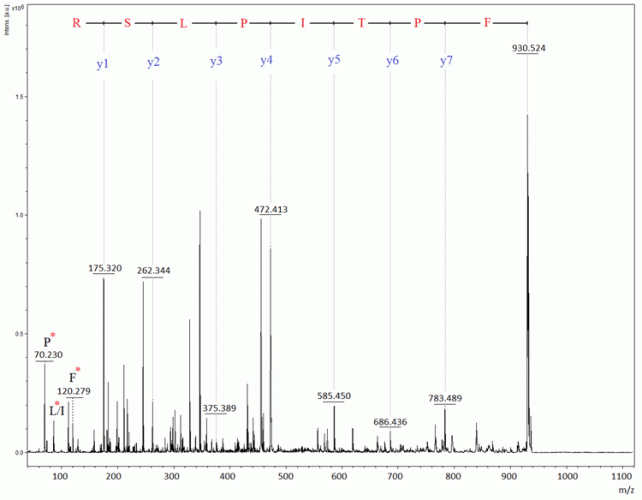
Figure 3: MALDI-PSD spectrum from a tryptic fragment at m/z 930.5 in the somatropin peptide map
It has also been suggested that reliable identification of proteins upon proteolytic cleavage is possible by a minimal set of experimentally-derived peptide masses; ie, signature peptides termed a minimal protein identifier (MPI) generated by MALDI-MS.18 Prior reports19 suggest that a minimum of four matching peptides and a sequence coverage of at least 20 percent is necessary for positive PMF-based protein identification.20 However, a fingerprint comprised of only two peptides has demonstrated to be sufficient for identifying a protein.21
A broad range of proteins and peptides, for various purposes of enhancement, can be obtained from the illicit market. The scope of this article is to describe identification of human growth hormone (hGH, somatropin) and delta sleep-inducing peptide (DSIP) and melanotan II by MALDI-TOF-MS, as described in Figure 1.
Identification of recombinant human growth hormone
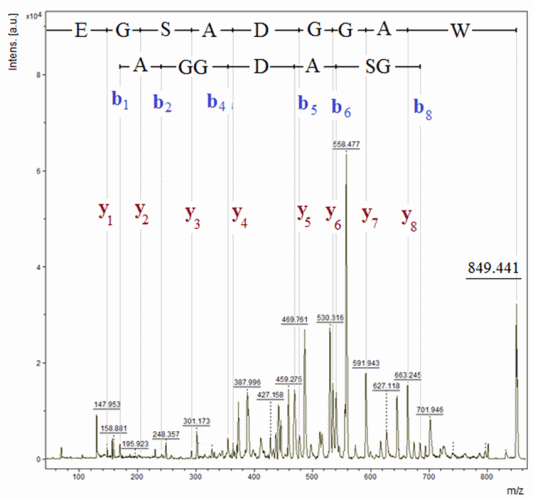
Figure 4: Amino acid sequencing of delta sleep-inducing peptide by MALDI-TOF-MS in PSD mode
Recombinant hGH, ie, somatropin, is a peptide hormone that consists of 191 amino acids with two disulfide bridges (Cys35–Cys165 and Cys182–Cys189) and promotes proteinogenesis and fat mobilisation and oxidation.22 Somatropin is a frequently occurring protein in illegally distributed samples seized by Swedish customs. Somatropin is identified through PMF (Figure 2).23
The spectrum from unalkylated tryptic somatropin, in Figure 2, contained a unique mass signal at m/z 1400.59, which originated from one fragment ion consisting of two tryptic peptides (ie, IVQCR and SVEGSCGF), being bound to each other through a disulphide bridge. This Cys (at position182)-Cys (at position189) cross-linked peptide fragment is used as a signature peptide for the identification. It is possible to select one or more peptides in the map to perform PSD analysis in order to confirm their amino acid sequences. As an example, Figure 3 demonstrates the PSD analysis of one of the tryptic peptides, ie, FPTLPEISR.
Mass signals originating from incomplete digestion fragments enhanced sequence coverage from 75 to 79 percent. A tryptic peptide containing oxidised methionine at site 170, ie, DMoxDKVETFLR, was also detected.
Identification of delta sleep-inducing peptide
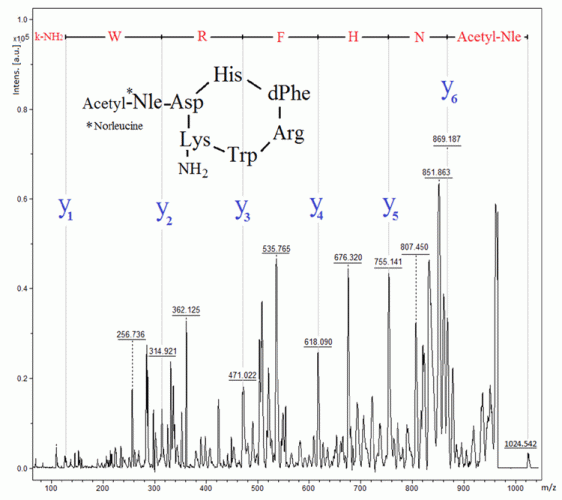
Figure 5: Identification of melanotan II by MALDI‑TOF-MS in PSD mode
The DSIP (WAGGDASGE; M [mono-isotopic] = 848.34 Da) was primarily believed to be involved in sleep regulation due to its apparent ability to induce slow-wave sleep in rabbits. However, it has been demonstrated that short-term treatment of chronic insomnia with DSIP is likely not of major therapeutic benefit.24 The peptide is marketed illegally, presumably for the treatment of insomnia, and is identified through the top-down approach by being directly exposed to the PSD analysis. The results summarised in Figure 4 confirmed the molecular mass as well as the primary structure of the peptide.
Identification of melanotan II
Melanotan II (Figure 5) is a cyclic peptide, which induces melanogensis (ie, tanning of the skin) by activation of the MC1 receptor, being an analogue to alfa melanocyte hormone (α-MSH).25 Melanotan activity on other receptors, ie, MC3 and MC4 receptors in the spinal cord and central nervous system, results in sexual arousal.25 However, injection of melanotan II can result in systemic toxicity and rhabdomyolysis.25 Similarly to DSIP, this peptide is also identified through the top-down approach by MALDI in PSD mode (Figure 5). It is to be noted that this polypeptide did not generate a useful MS-MS spectrum when analysed by LC-MS.26
Conclusions
The quality and safety of illegally distributed products, with respect to the purity and endotoxin level as well as the microbiological quality, may present a serious health threat to the users and to public health. MALDI-TOF-MS provides an efficient procedure for the identification of peptides and proteins in illegally distributed samples. The use of trypsin as a proteolytic enzyme generated peptide fragments covering 40 to 80 percent of the amino acid sequence of the analysed human growth hormone. The presence of a signature peptide in the peptide map expedited analyte identification considerably. MALDI-TOF-MS was also applied in the PSD mode for the amino acid sequencing of selected tryptic peptides as well as small peptides.
Author Biographies

TORGNY RUNDLÖF is a pharmaceutical evaluator at the Swedish MPA. He received his PhD in organic chemistry in 1998 at Stockholm University. The study, which was carried out with Professor Göran Widmalm, focused on NMR studies of the structure and dynamics of carbohydrates. In 2000 he joined the MPA as an NMR specialist. His recent work deals with supervision of various registered medicinal products, using spectroscopy techniques such as NMR, IR and MS, some of which has been published. Currently, he examines the potential of NMR and MS for characterisation of biopharmaceuticals including illegally distributed products.
 AHMAD AMINI is a pharmaceutical assessor at the Swedish MPA. He received his PhD in analytical pharmaceutical chemistry in 1998 at Uppsala University. The thesis work, which involved enantiomeric separation of pharmaceutical compounds using CE, was carried out under supervision of Professor Douglas Westerlund and Associate Professor Curt Pettersson. Following post-doctoral studies on proteomics at Purdue University, West Lafayette, US, under supervision of Professor Fred E. Regnier he joined the MPA laboratory in 2001. Currently, he is involved in method development for the European Pharmacopoeia and MS characterisation of biopharmaceuticals including illegally distributed products.
AHMAD AMINI is a pharmaceutical assessor at the Swedish MPA. He received his PhD in analytical pharmaceutical chemistry in 1998 at Uppsala University. The thesis work, which involved enantiomeric separation of pharmaceutical compounds using CE, was carried out under supervision of Professor Douglas Westerlund and Associate Professor Curt Pettersson. Following post-doctoral studies on proteomics at Purdue University, West Lafayette, US, under supervision of Professor Fred E. Regnier he joined the MPA laboratory in 2001. Currently, he is involved in method development for the European Pharmacopoeia and MS characterisation of biopharmaceuticals including illegally distributed products.
 HENRIK LODÉN is a pharmaceutical evaluator at the Swedish MPA. He received his PhD in analytical pharmaceutical chemistry in 2008 at Uppsala University. The thesis work, which involved separation of pharmaceutical compounds using different modes of CE, was carried out under supervision of Professor Curt Pettersson, Associate Professor Torbjörn Arvidsson and Associate Professor Ahmad Amini. Following employments in the pharmaceutical industry and as a researcher in academia, using MALDI and LC-MS, he joined the MPA laboratory in 2015. Currently, he is involved in method development for the European Pharmacopoeia and MALDI identification of illegally distributed biopharmaceutical products.
HENRIK LODÉN is a pharmaceutical evaluator at the Swedish MPA. He received his PhD in analytical pharmaceutical chemistry in 2008 at Uppsala University. The thesis work, which involved separation of pharmaceutical compounds using different modes of CE, was carried out under supervision of Professor Curt Pettersson, Associate Professor Torbjörn Arvidsson and Associate Professor Ahmad Amini. Following employments in the pharmaceutical industry and as a researcher in academia, using MALDI and LC-MS, he joined the MPA laboratory in 2015. Currently, he is involved in method development for the European Pharmacopoeia and MALDI identification of illegally distributed biopharmaceutical products.
References
1. Karas M, Hillenkamp F. Anal Chem 1988, 60, 2299–3201.
2. Pappin DJC, Hojrup P, Bleasby AJ. Current Biol 1993, 3, 327–332.
3. Gevaert K, Vandekerckhove J. Electrophoresis 2000, 21, 1145–1154.
4. Pandey A, Mann M. Nature 2000, 405, 837–846.
5. Amini A, Dormady SJ, Riggs L, Regnier FE. J Chromatogr A 2000, 894, 345–355.
6. Spengler J. Mass Spectrom 1997, 32, 1019–1036.
7. Cleveland DW, Fischer SG, Kirschner MW, Laemmli UK. J Biol Chem 1977, 252, 1102–1106.
8. Matsudaira P. J Biol Chem 1987, 262, 10035–10038.
9. Domon B, Aebersold R. Science 2006, 312, 212–217.
10. Aebersold R, Mann M. Nature 2003,422:198–207.
11 Yates III JR, Speicher S, Griffin PR, Hunkapiller T, Anal Biochem, 1993, 214, 397–408.
12. Wettenberg A, Organ AJ, Schneider K, Tyldesley R, Bordoli R, Bateman RH, J Am Soc Mass Spectrom 2002,
13, 772–783. 13. Zhang WZ, Chait BT. Anal Chem 2000, 72, 2482–2489.
14. Perkins DN, Pappin DJC, Creasy JM, Cottrell JS. Electrophoresis 1999, 20, 3551–3567.
15. Eng JK, Mccormack AL, Yates JR. J Am Soc Mass Spectrom 1994, 5, 976–989.
16. Damodaran S, Wood TD, Nagarajan P, Rabin RA. Geno Prot Bioinfo 2007, 5, 152–157.
17. Thevis M, Schänzer W. Anal Bioanal Chem 2007, 388, 1351–1358.
18. Ji J, Chakraborty A, Geng M, Zhang X, Amini A, Bina M, Regnier FE. J Chromatogr B 2000, 745, 197–210.
19. Chamrad DC, Körting G, Stühler K, Meyer HE, Klose J, Blüggel M. Proteomics 2004, 4, 619–628.
20. Biron DG, Brun C, Lefevre T, Lebarbenchon C, Loxdale HD, Chevenet F, Brizard JP, Thomas F. Proteomics 2006, 6, 5577–5596.
21. Henzel WJ, Watanabe C. J Am Soc Mass Spectrom 2003, 14, 931–942.
22. Hober FF, Haymond MW. J Clin Invest 1990, 86, 265–272.
23. McCuskey R, Urbaschek R, Urbaschek B. Cardiovasc Res 1996, 32, 752–763.
24. Schoenenberger GA, Maier PF, Tobler HJ, Monnier M. Eur J Phys 1977, 369, 99–109.
25. Nelson M E, Bryant S M, Aks S E. Clin Toxicol (Phila) 2012, 50, 1169–1173.
26. Breindahl H, Evans-Brown M, Hidersson P, McVeigh J, Bellis M., Stensballe A, Kimergård A. Drug Test Analysis 2015, 7, 164–172.
Issue
Related topics
Analytical techniques, Drug Counterfeiting, Mass Spectrometry, Proteins





Well written and to the point. I appreciate the detail in this article!