

The importance of being small: miniaturisation of freeze drying equipment
Posted: 18 August 2017 | Henning Gieseler, Margit Gieseler | No comments yet
Developmental activities for freeze-dried products are dictated by the limited availability and high cost of newly developed active compounds, and by Quality-by-Design requirements. Laboratory-scale freeze dryers used for formulation and process development show an excessive variety of designs and instrumentation concepts, making scale-up activities a challenge. The development of miniaturised equipment may provide enormous benefits in terms of development times and costs.
Pharmaceutical drug product development includes processing at different batch-size levels (scales), depending on the stage of the development process and the number of samples required for clinical trials, stability studies, and market supply. In general, these batch sizes are classified into laboratory, pilot, and production scale.1 For freeze-dried products, process development and optimisation is typically done using laboratory-scale freeze dryers of approx. 0.1-0.5m2 shelf area. Pilot-scale freeze dryers of up to approximately 5m2 shelf area are used to manufacture material for clinical testing and stability samples, and may also allow cycle adaptations for further process optimisation. For market supply, production-scale freeze dryers with shelf areas up to 50m2 may be used.
The freeze-drying process consists of three phases: first, the freezing step; second, the primary drying (or main drying) phase; third – secondary drying.2 Proper control and monitoring of all three phases is mandatory to ensure drug product quality complies with the defined Critical Quality Attribute (CQA) limits. During freezing, the solvent is converted into a solid, with freezing conditions defining the ice network (ice crystal size and shape) formed within the frozen product solution. Since ice crystals leave pores of the same size and shape upon sublimation, the inner cake morphology is also defined during the freezing phase.2 The second phase of a freeze-drying process – primary drying – is governed by sublimation of the frozen solvent. This phase represents the longest part of the overall process.3 Accordingly, it is most relevant if cycle optimisation is desired. During secondary drying, unfrozen water is removed from the product by diffusion and desorption, resulting in residual moisture contents of typically 1% or less, depending on the process conditions used as well as on other factors such as the nature of the formulation.4,5
During development, the freeze-drying process should balance economical aspects (i.e., short process times) and the formulation’s ‘needs’, such as the maximum allowable product temperature during primary drying, i.e., the onset of collapse temperature (Toc),6 or an optimum residual moisture range for stability.7,8
Obstacles in scale-up
Heat and mass transfer determines the success of a freeze-drying cycle at given process conditions. Heat transfer into the product is based on three different pathways: first, direct conduction at points of contact between the container system (vial) and the temperature-controlled shelf; second, heat transfer by gas conduction; and third, by radiation.9 The relative contribution of these pathways and effective heat transmitted is dependent on factors such as the container system (eg, molded or tubing vial),10 chamber pressure and shelf temperature setting,10 freeze dryer design and capacity,11 and loading conditions.12 All these factors determine the product temperature over time profile, which first needs to be optimised for the particular formulation during development, and then transferred during scale-up.
The initial scale-up step from laboratory to pilot scale includes the biggest changes regarding equipment design and performance, as well as environmental conditions. Accordingly, the cycle developed on the laboratory-scale freeze dryer may not be run on a 1:1 basis with pilot scale equipment. For example, laboratory-scale freeze dryers are typically equipped with acrylic chamber doors, which contribute to increased radiation heat transfer, resulting in pronounced edge vial effects.13 In addition, the environment changes as the process moves from the laboratory, containing airborne particles, to sterile conditions at pilot or production scale. Since airborne particles are a source of nucleation, the degree of super-cooling (difference between temperature when ice crystal formation starts and thermodynamic freezing point) of a product solution is much higher in pilot or production scale compared to laboratory scale. This causes small ice crystals to form, which imposes higher resistance to water vapour flow and thus affects the freeze-drying process with regard to product temperature during primary drying, primary drying time, and residual moisture content.14 That is, the product temperature profile is likely to change. To overcome this major scale-up issue, several recently-suggested technologies allow control of nucleation, ie, start of ice crystal formation in all vials at the same time and the same desired product temperature.15
To be useful, however, such technologies must be applicable at all scales. To account for scale-up factors related to freeze-drying equipment design and capacity, the equipment must be thoroughly characterised. Operational qualification protocols testing the performance and impact of all relevant components under load help to identify critical issues. The relevant components include condenser (maximum deposition rate, maximum total load), shelves (cooling and heating rates, homogeneity of shelf surface temperature), chamber design (edge vial effect, shelf interdistance), and controls (sensor types, achievable control levels).16,17 Sublimation tests, shelf temperature mapping, and emissivity measurements are typically conducted to calculate relevant parameters based on heat and mass transfer equations, eg, heat transfer coefficients, resistances, radiative heat transfer, product temperatures, etc.16-18
Recent scale-down approaches
Limited availability of active pharmaceutical ingredient (API) during early phases of development, high API costs, and typically limited access to pilot or production-scale equipment for technical batches require most developmental activities to be conducted at laboratory scale. Efforts have recently been made to further scale-down equipment that allows characterisation of the formulation’s performance during freezing and drying in single vials, or provide very small batches for, for example, formulation-finding studies. Based on the above-mentioned challenges that come with scale-up, the design of such small-scale equipment must (1) reflect the drying conditions in larger scale equipment, and (2) allow appropriate process control and monitoring to be suitable for smooth transfer of the developed cycle.
It may be argued that standard laboratory-scale freeze dryers can also be used for processing single vials or small batches by partial loading, or by simultaneous processing of different formulations. For formulation-screening studies this may be valid since typically conservative process conditions are used, based on the thermal properties of the worst-case formulation, to exclude potential impact of the drying conditions on CQAs and stability. However, for process development and optimisation activities, both partial loading and the processing of different formulations in the same batch may lead to non-representative process and product results. As examples for ‘non-representative’, the premature detection of primary drying endpoints and misleading results for residual moisture content after secondary drying could be listed.12
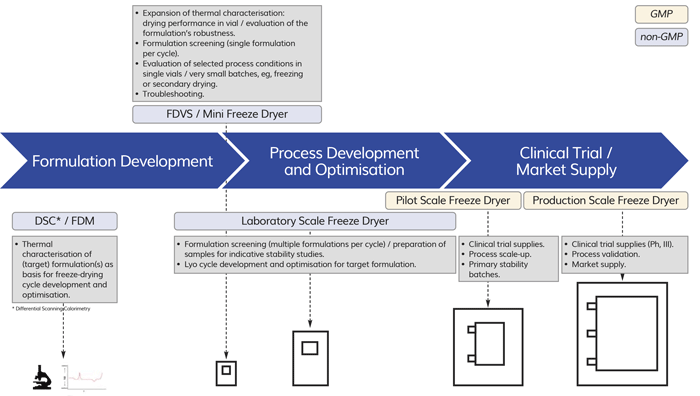
Figure 1: Overview of different objectives in freeze-dried drug product development, including scale classification.
The idea of a mini-scale freeze dryer adaptable to process conditions present in larger scale equipment may therefore be cost and time saving, eg, if selected process conditions can be evaluated in few vials and in parallel to other laboratory-scale runs. Figure 1 illustrates an overview on the different objectives during product development and the typical scales used, including a potential fit for miniaturised freeze-drying equipment into this scheme. In the following sections, examples of the most recent developments of mini-scale equipment are reviewed and further discussed.
Freeze-drying vial system (FDVS)
The current state-of-the-art technology to determine the collapse temperature of a formulation is light transmission freeze dry microscopy (LT-FDM),19 simply known as freeze dry microscopy (FDM).6 This allows the starting point of viscous flow in the dried matrix next to the sublimation interface to be determined at a pore-size level, ie, Toc. The FDVS (Linkam Scientific, Tadworth, UK) is a new system which can be used to observe the product during the freeze-drying process in a standard container system. The equipment consists of a chamber capable of loading up to seven vials (2R) or one vial (>2R), equipped with Pirani and capacitance manometer, liquid nitrogen condenser and shelf cooling, thermocouples for product temperature measurement, an optical system (Nikon) controllable in X, Y, and Z axis, including a 10x darkfield lens to visualise the drying behaviour, and software-supported controls and monitoring capabilities.19
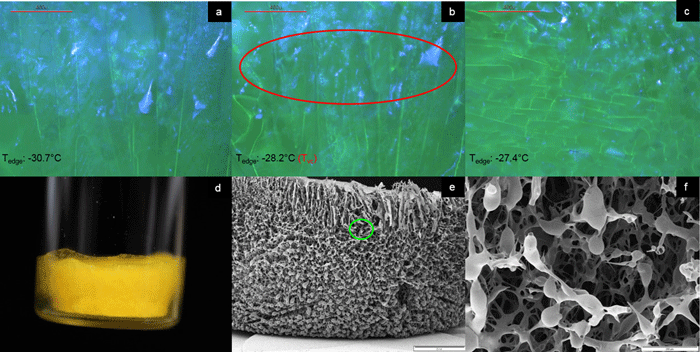
Figure 2: Illustration of collapse detection using the FDVS system (a-c), the macroscopic (d), and the microscopic appearance at magnification 10x (e) and 100x (f) of a sample freeze dried in a FDVS system. Sample: sucrose 50 mg/mL + fluorescein 1 mg, 3 mL fill in 10R TopLyo vial, chamber pressure 200 mTorr. Data from Ref. 19.
Experimental data suggests that structural changes can be detected in an FDVS at temperatures not equal to Toc obtained by FDM. Therefore, the critical temperature obtained by FDVS was defined as Tvc (onset of collapse temperature in a vial).19 Figures 2 (a-c) illustrates the drying performance and Tvc determination for sucrose 50mg/mL including 1mg of fluorescein in an FDVS. During analysis, the sample was illuminated with UV light to better visualise the events. In (a), the primary drying progress is shown, with the dried matrix appearing mainly blue. On collapsing of the structure, the blue colour disappears and the pore structure widens (b). For this sample, Tvc was detected at -28.2°C, as indicated by a thermocouple positioned close to the area of observation (Tedge). Picture (c) illustrates the ongoing deterioration of the matrix, with the blue colour almost gone. It must be noted that Toc measured by FDM for this particular sample was -32.6°C (pictures not shown), which is in agreement with data obtained earlier for sucrose 50mg/mL.6 FDVS data of the investigated sucrose formulation agree with data obtained for 5% sucrose using another optical technology recently introduced to observe structural integrity during drying in a container system – optical coherence tomography-based FDM (OCT-FDM).20 Figures 2 (d-f) show the macroscopic and microscopic appearance of the freeze-dried cake. Macroscopically, some shrinkage could be observed, which was especially pronounced in the bottom area of the cake (d). SEM pictures show an overall intact cake with droplet-like structures at the edge (e). Generally, droplet-like structures can be attributed to (micro)collapse, caused by viscous flow. Higher magnification of the area enclosed by the green circle in (e) confirms this observation (f).
The data obtained provide useful information about the robustness of the formulation, and the FDVS may therefore be used to study the freezing and collapse behaviour in standard container systems.19 Since the complete freeze-drying process can be simulated, it may also be used for indicative freeze-drying studies with low material consumption, or for evaluation of heat transfer characteristics of container systems.19 Current demerits of the FDVS are rather high radiation effects, which are expected to impact the freezing and drying behaviour of the product, limited observation depth (only the sample area close to the vial wall can be visualised), and limited visualisation options for non-transparent container systems.19
Mini freeze dryers
Newly developed mini freeze dryers such as the LyoCapsule (SP Scientific, US) or the MicroFD (Millrock Systems, US) differ in several aspects from standard small-scale freeze dryers. For example, by the available shelf area, taking up to 7 20R (LyoCapsule)21 or 19 10R (MicroFD)22 vials. Similar to standard small-scale freeze dryers, pronounced radiation is expected in such miniaturised systems, which significantly impacts heat transfer and results in atypical drying conditions as discussed above. Accordingly, a major objective when developing equipment capable of providing scalable cycles is to create freeze-drying conditions that reflect the situation in larger systems.
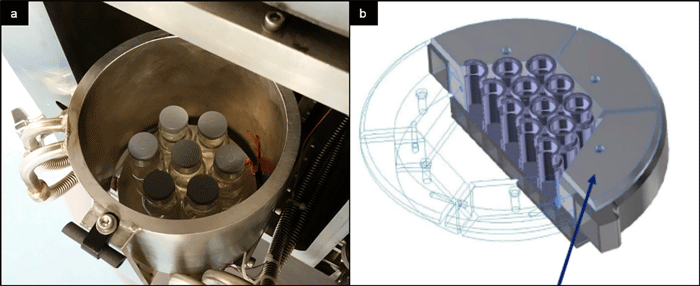
Figure 3: Shielding of vial array in newly developed mini freeze dryers. (a): cylindrical temperature-controlled wall in LyoCapsule. Picture courtesy of SP Scientific. (b) thermal conductor segments LyoSim in MicroFD.^22
To overcome the issue of atypical drying conditions, shielding of the vial array in the form of temperature-controlled chamber walls (LyoCapsule)^21, or segments of thermal conductors surrounding the vials (LyoSim in MicroFD)22 may be used, cf Figure 3 . Such set-up not only positively impacts the atypical drying situation, but also allows heat transfer to be modified by adjusting the wall or conductor element temperature to match with the conditions present in a larger freeze dryer. The parameter reflecting the amount of heat transmitted for a particular vial system is Kv (vial heat transfer coefficient).^10 Kv is a crucial parameter in steady-state heat and mass transfer equations used to calculate process-related parameters, for example, the target shelf temperature required to obtain a certain product temperature over time profile.
First experiments show promising results. By setting the wall temperature to -33°C (equalling the product temperature of the center vial in the LyoCapsule), Kv values of the vial array could be adapted to the values obtained for centre vials in a larger development freeze dryer.23 Similar options with regard to Kv modification were reported for setting the LyoSim temperature to either slightly lower or higher values than the product temperature.22 Another relevant difference to standard small-scale development dryers relates to capacity and instrumentation. A mini freeze dryer intended to simulate drying conditions in larger systems needs to be well-equipped with regards to process control and monitoring tools. Standard should be larger scale-comparable design (eg, 316L stainless steel for shelves and chamber), appropriate refrigeration capacity, several product temperature probes, comparative pressure measurement (Pirani/capacitance manometer endpoint detection), and software-supported controls. In addition, different process analytical technology (PAT) tools available on larger freeze dryers such as SMART/MTM, TDLAS (tunable laser diode laser absorption spectroscopy), or ControLyo (LyoCapsule),21 or AccuFlux/LyoPAT, or FreezeBooster (MicroFD)22 should be included for optimal data comparison.
Conclusion
Miniaturisation of freeze-drying equipment provides the opportunity to gain useful product and process information by reduced consumption of material and time. A prerequisite for scale-up success is an appropriate design of such mini freeze-drying systems that must ensure process conditions comparable to larger equipment. Different systems have been developed and are currently tested. Overall, it still seems a long way to go until a scientifically coherent and standardised scale-up procedure has been developed and introduced in pharmaceutical industry. However, first promising results have paved the way into the right direction.
BIOGRAPHIES
Dr Henning Gieseler is the group leader of the ‘Freeze Drying Focus Group’ at the FAU in Erlangen and Chief Scientific Officer at GILYOS in Wuerzburg, Germany. His research interest include scale-up, process analytical technology (PAT) and primary packaging materials applied in pharmaceutical freeze drying. * Corresponding author: [email protected]
Dr Margit Gieseler Chief Executive Officer at GILYOS, a freeze-drying service provider for the pharmaceutical industry. She has been a licensed pharmacist in Germany since 2001, and received her PhD from the Department of Pharmaceutics, University of Wuerzburg.
REFERENCES
- McQuaid J, Long CP. Strategic Approaches to Process Optimization and Scale-up. Pharm. Tech. 2010;34(9). pharmtech.com/print/213454?page=full. Link accessed June 2017.
- Pikal Freeze Drying. In: J. Swarbrick (Ed.), Encyclopedia of Pharmaceutical Technology. 2007; pp.1807-1833.
- Nail SL, Pikal MJ. The Fundamentals of Freeze Drying. Short Course. Freeze Drying of Pharmaceuticals & Biologicals Conference, August 6, 2008, Great Divide Lodge Breckenridge, Colorado.
- Pikal MJ, Shah S. Intravial Distribution of Moisture During the Secondary Drying Stage of Freeze Drying. PDA J. Pharm. Sci. Technol. 1997;51(1):17-24.
- Pikal MJ, Shah S, Roy ML, Putman R. The secondary drying stage of freeze drying: drying kinetics as a function of temperature and chamber pressure. Int. J. of Pharm. 1990;60:203-217.
- Meister E, Gieseler H. Freeze-Dry Microscopy of Protein/Sugar Mixtures: Drying Behavior, Interpretation of Collapse Temperatures and a Comparison to Corresponding Glass Transition Data. J. Pharm. Sci. 2009;98(9):3072-3087.
- Greiff D. Protein structure and freeze drying: The effects of residual moisture and gases. Cryobiology. 1971;8:145-152.
- Breen ED, Curley JG, Overcashier DE, Hsu CC, Shire SJ. Effect of Moisture on the Stability of a Lyophilized Humanized Monoclonal Antibody Formulation. Pharm. Res. 2001;18(9):1345-1353.
- Pikal MJ, Roy ML, Shah S. Mass and Heat Transfer in Vial Freeze-Drying of Pharmaceuticals: Role of the Vial. J. Pharm. Sci. 1984;73(9):1224-1237.
- Hibler S, Wagner CH, Gieseler H. Vial Freeze-Drying, Part 1: New Insights into Heat Transfer Characteristics of Tubing and Molded Vials. Pharm. Sci. 2012;101(3):1189-1201.
- Tsinontides SC, Rajniak P, Pham D, Hunke WA, Placek J, Reynolds SD. Freeze drying – principles and practice for successful scale-up to manufacturing. Int. J. Pharm. 2004;280:1-16.
- Patel S, Jameel F, Pikal MJ. The Effect of Dryer Load on Freeze Drying Process Design. J. Pharm. Sci. 2010;99(10):4363-4379.
- Rambhatla S, Pikal MJ. Heat and Mass Transfer Scale-up Issues During Freeze-Drying, I: Atypical Radiation and the Edge Vial Effect. AAPS PharmSciTech. 2003;4(2):111-120.
- Rambhatla S, Ramot R, Bhugra C, Pikal MJ. Heat and Mass Transfer Scale-up Issues During Freeze-Drying, II: Control and Characterization of the Degree of Supercooling. AAPS PharmSciTech. 2004;5(4):1-9.
- Gieseler H, Stärtzel P. Controlled Nucleation in Freeze-drying. Pharm. Rev. 2012;17(5):63-68.
- Rambhatla S, Tchessalov S, Pica MJ. Heat and Mass Transfer Scale-up Issues During Freeze-Drying, III: Control and Characterization of Dryer Differences via Operational Qualification Tests. AAPS PharmSciTech. 2006;7(2).Article 39.
- Mayeresse Y. Challenges and Opportunities when Scaling up your Lyophilized Pharmaceutical Products. Presentation, GSIA Educational Course, Basel, 2013.
- Tchessalov S. Lyophilization: cycle robustness and process tolerances, transfer and scale up. www.europeanpharmaceuticalreview.com/article/1263/lyophilization-cycle-robustness-and-process-tolerances-transfer-and-scale-up/. Link accessed June 2017.
- Huang Z. New Development to Assess the Critical Formulation Temperature: Light Transmission Freeze Dry Microscopy (LT-FDM) vs. Freeze Drying Vial System (FDVS). / M. Schwertner, D. Stacey. Freeze Drying Vial System (FDVS) – Features, modular design and upcoming extensions. Presentation at Lyoscopedia 2017, 30 March.
- Greco K, Mujat M, Galbany-Kinney KL, Hammer DX, Ferguson RD, Iftimia N, Mulhall P, Sharma P, Kessler WJ, Pikal MJ. Accurate Prediction of Collapse Temperature using Optical Coherence Tomography-Based Freeze-Drying Microscopy. J. Pharm. Sci. 2013;102(6):1773-1785.
- Yusoff Z. Scientific Advancing Lyophilization. Presentation given at CPPR, Freeze Drying of Pharmaceuticals & Biologicals Short Course & Conference. July 12-15, 2016, Breckenridge, Colorado, USA.
- Thompson TN, Wang Q, Reiter C. Developing Transferable Freeze Drying Protocols Using Accuflux and a MicroFD. Presentation given at Peptalk 2017, San Diego, USA. Downloaded from https://pharmahub.org/groups/lyo/tools, link accessed June 2017.
- Obeidat W, Sahni E, Grobelny P, Kessler W, Pikal M. Development of a Mini-Freeze Dryer for Pharmaceutical Product Formulation and Process Development. Poster presentation, AAPS National Biotechnology Conference, May 16-18, 2016. Boston, Massachusetts, USA.
Issue
Related topics
Analytical techniques, Drug Development, Freeze Drying, Lyophilisation





